

Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model
- PMID: 19088194
- PMCID: PMC2603435
- DOI: 10.1073/pnas.0807121105
Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model
Abstract
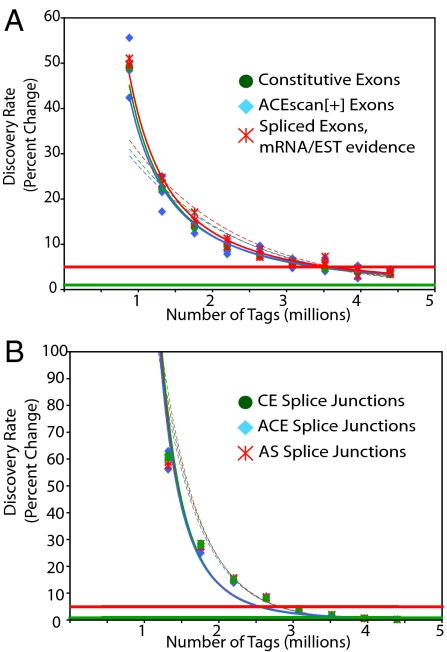
High-throughput sequencing has rapidly gained popularity for transcriptome analysis in mammalian cells because of its ability to generate digital and quantitative information on annotated genes and to detect transcripts and mRNA isoforms. Here, we described a double-random priming method for deep sequencing to profile double poly(A)-selected RNA from LNCaP cells before and after androgen stimulation. From approximately 20 million sequence tags, we uncovered 71% of annotated genes and identified hormone-regulated gene expression events that are highly correlated with quantitative real time PCR measurement. A fraction of the sequence tags were mapped to constitutive and alternative splicing events to detect known and new mRNA isoforms expressed in the cell. Finally, curve fitting was used to estimate the number of tags necessary to reach a "saturating" discovery rate among individual applications. This study provides a general guide for analysis of gene expression and alternative splicing by deep sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Cheng J, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. - PubMed
-
- Kapranov P, et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. - PubMed
-
- Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8:413–423. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
