

Toll-like receptors, wound healing, and carcinogenesis
- PMID: 19089397
- PMCID: PMC2791674
- DOI: 10.1007/s00109-008-0426-z
Toll-like receptors, wound healing, and carcinogenesis
Abstract
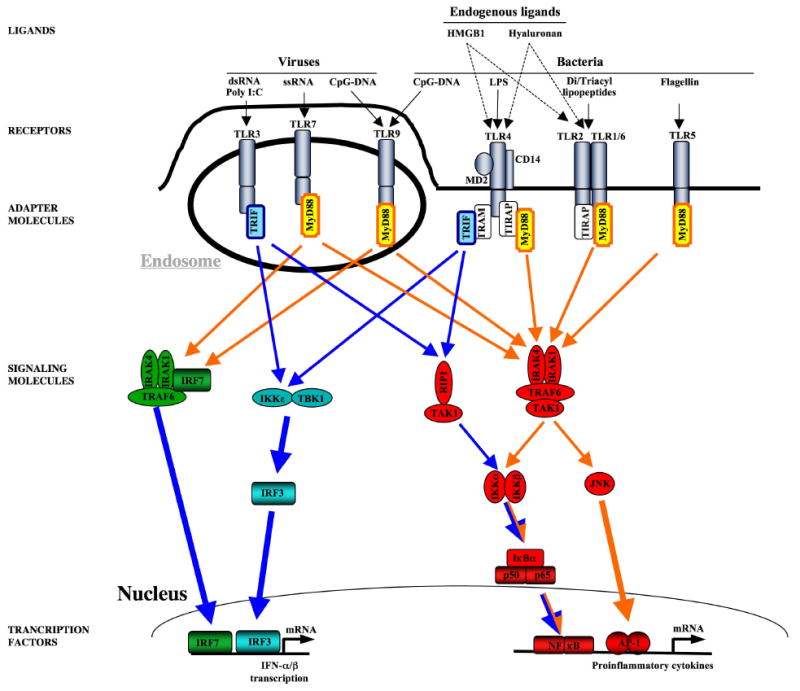
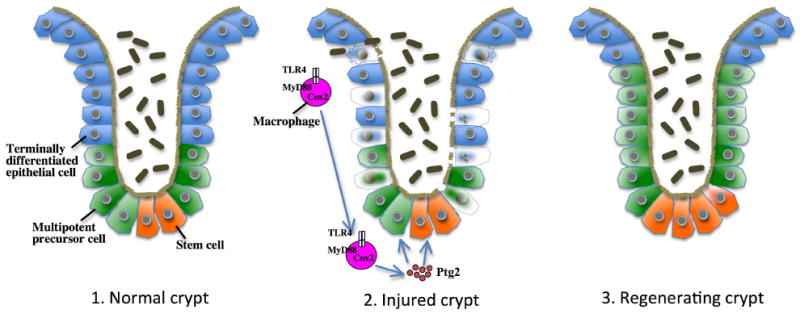
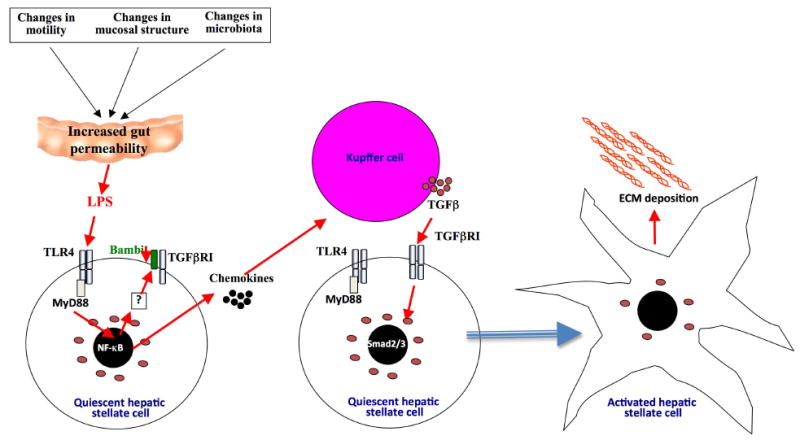
Following acute injury, the concerted action of resident and nonresident cell populations evokes wound healing responses that entail a temporary increase in inflammation, extracellular matrix production, and proliferation to ultimately restore normal organ architecture. However, chronic injury evokes a perpetuating wound healing response promoting the development of fibrosis, organ failure, and cancer. Recent evidence points toward toll-like receptors (TLRs) as important regulators of inflammatory signals in wound healing. Here, we will review the activation of TLRs by different endogenous and bacterial TLR ligands during wound healing, and the contribution of TLR-induced signals to injury, fibrogenesis, regeneration, and carcinogenesis. We will discuss the hypothesis that TLRs act as sensors of danger signals in injured tissue to switch the wound healing response toward fibrogenesis and regeneration as a protective response to imminent danger at the cost of an increased long-term risk of developing scars and cancer.
Figures
References
-
- Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE, Jr, Flaherty KR, Schwartz DA, Noble PW, Raghu G, Brown KK. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:963–967. DOI 142/12_Part_1/963 [pii] - PubMed
-
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. - PubMed
-
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
