

Loss of translation elongation factor (eEF1A2) expression in vivo differentiates between Wallerian degeneration and dying-back neuronal pathology
- PMID: 19094180
- PMCID: PMC2666133
- DOI: 10.1111/j.1469-7580.2008.01007.x
Loss of translation elongation factor (eEF1A2) expression in vivo differentiates between Wallerian degeneration and dying-back neuronal pathology
Abstract
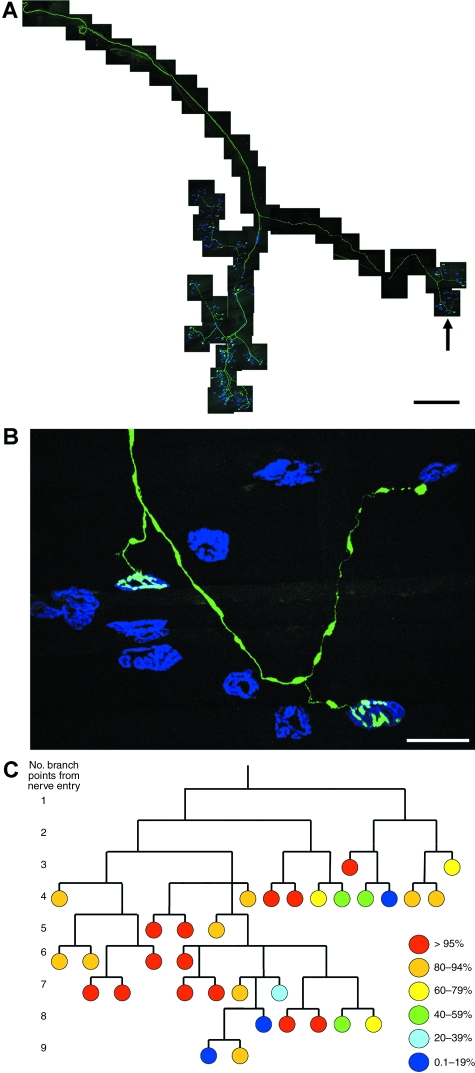
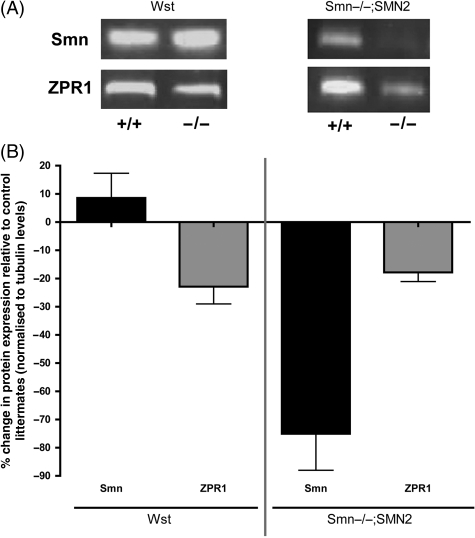
Wallerian degeneration and dying-back pathology are two well-known cellular pathways capable of regulating the breakdown and loss of axonal and synaptic compartments of neurons in vivo. However, the underlying mechanisms and molecular triggers of these pathways remain elusive. Here, we show that loss of translation elongation factor eEF1A2 expression in lower motor neurons and skeletal muscle fibres in homozygous Wasted mice triggered a dying-back neuropathy. Synaptic loss at the neuromuscular junction occurred in advance of axonal pathology and by a mechanism morphologically distinct from Wallerian degeneration. Dying-back pathology in Wasted mice was accompanied by reduced expression levels of the zinc finger protein ZPR1, as found in other dying-back neuropathies such as spinal muscular atrophy. Surprisingly, experimental nerve lesion revealed that Wallerian degeneration was significantly delayed in homozygous Wasted mice; morphological assessment revealed that approximately 80% of neuromuscular junctions in deep lumbrical muscles at 24 h and approximately 50% at 48 h had retained motor nerve terminals following tibial nerve lesion. This was in contrast to wild-type and heterozygous Wasted mice where < 5% of neuromuscular junctions had retained motor nerve terminals at 24 h post-lesion. These data show that eEF1A2 expression is required to prevent the initiation of dying-back pathology at the neuromuscular junction in vivo. In contrast, loss of eEF1A2 expression significantly inhibited the initiation and progression of Wallerian degeneration in vivo. We conclude that loss of eEF1A2 expression distinguishes mechanisms underlying dying-back pathology from those responsible for Wallerian degeneration in vivo and suggest that eEF1A2-dependent cascades may provide novel molecular targets to manipulate neurodegenerative pathways in lower motor neurons.
Figures





Similar articles
-
The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice.Brain. 2005 Feb;128(Pt 2):405-16. doi: 10.1093/brain/awh368. Epub 2005 Jan 11. Brain. 2005. PMID: 15644421
-
Rapid loss of motor nerve terminals following hypoxia-reperfusion injury occurs via mechanisms distinct from classic Wallerian degeneration.J Anat. 2008 Jun;212(6):827-35. doi: 10.1111/j.1469-7580.2008.00909.x. J Anat. 2008. PMID: 18510509 Free PMC article.
-
Progressive loss of motor neuron function in wasted mice: effects of a spontaneous null mutation in the gene for the eEF1 A2 translation factor.J Neuropathol Exp Neurol. 2005 Apr;64(4):295-303. doi: 10.1093/jnen/64.4.295. J Neuropathol Exp Neurol. 2005. PMID: 15835265
-
The relationship of neuromuscular synapse elimination to synaptic degeneration and pathology: insights from WldS and other mutant mice.J Neurocytol. 2003 Jun-Sep;32(5-8):863-81. doi: 10.1023/B:NEUR.0000020629.51673.f5. J Neurocytol. 2003. PMID: 15034273 Review.
-
Programmed axon death, synaptic dysfunction and the ubiquitin proteasome system.Curr Drug Targets CNS Neurol Disord. 2004 Jun;3(3):227-38. doi: 10.2174/1568007043337436. Curr Drug Targets CNS Neurol Disord. 2004. PMID: 15180483 Review.
Cited by
-
SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy.Hum Mol Genet. 2010 Nov 1;19(21):4216-28. doi: 10.1093/hmg/ddq340. Epub 2010 Aug 12. Hum Mol Genet. 2010. PMID: 20705736 Free PMC article.
-
Morphological analysis of neuromuscular junction development and degeneration in rodent lumbrical muscles.J Neurosci Methods. 2014 Apr 30;227:159-65. doi: 10.1016/j.jneumeth.2014.02.005. Epub 2014 Feb 14. J Neurosci Methods. 2014. PMID: 24530702 Free PMC article.
-
Analysis of the Expression and Subcellular Distribution of eEF1A1 and eEF1A2 mRNAs during Neurodevelopment.Cells. 2022 Jun 9;11(12):1877. doi: 10.3390/cells11121877. Cells. 2022. PMID: 35741005 Free PMC article.
-
Induction of cell stress in neurons from transgenic mice expressing yellow fluorescent protein: implications for neurodegeneration research.PLoS One. 2011 Mar 8;6(3):e17639. doi: 10.1371/journal.pone.0017639. PLoS One. 2011. PMID: 21408118 Free PMC article.
-
Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy.PLoS Genet. 2009 Dec;5(12):e1000773. doi: 10.1371/journal.pgen.1000773. Epub 2009 Dec 18. PLoS Genet. 2009. PMID: 20019802 Free PMC article.
References
-
- Balice-Gordon RJ, Smith DB, Goldman J, et al. Functional motor unit failure precedes neuromuscular degeneration in canine motor neuron disease. Ann Neurol. 2000;47:596–605. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
