

A genome assembly-integrated dog 1 Mb BAC microarray: a cytogenetic resource for canine cancer studies and comparative genomic analysis
- PMID: 19096206
- PMCID: PMC2874680
- DOI: 10.1159/000163088
A genome assembly-integrated dog 1 Mb BAC microarray: a cytogenetic resource for canine cancer studies and comparative genomic analysis
Abstract
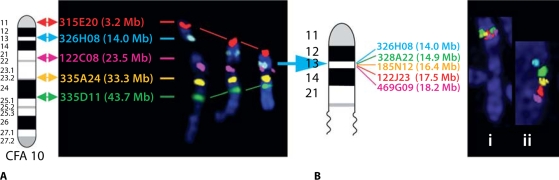
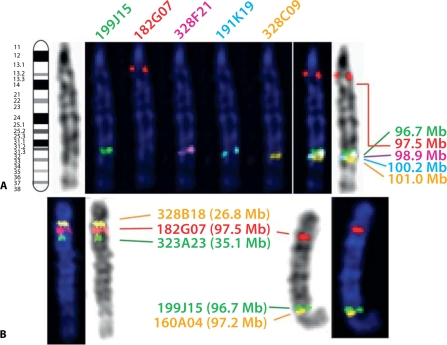
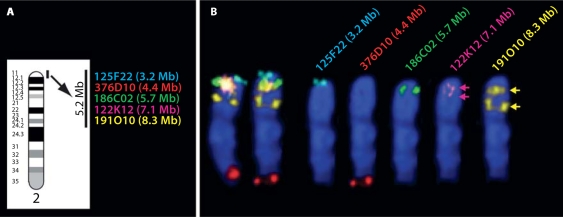
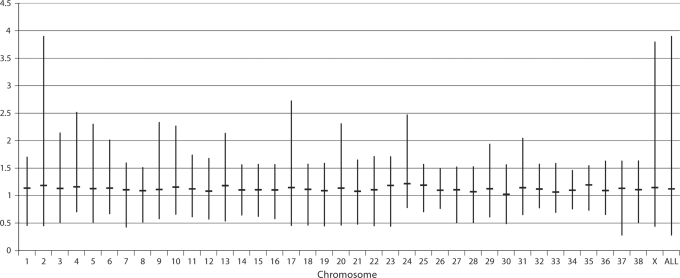
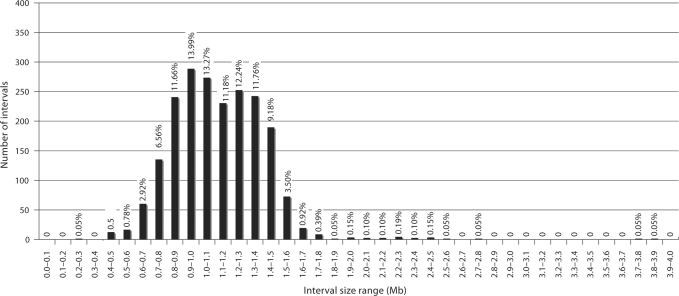
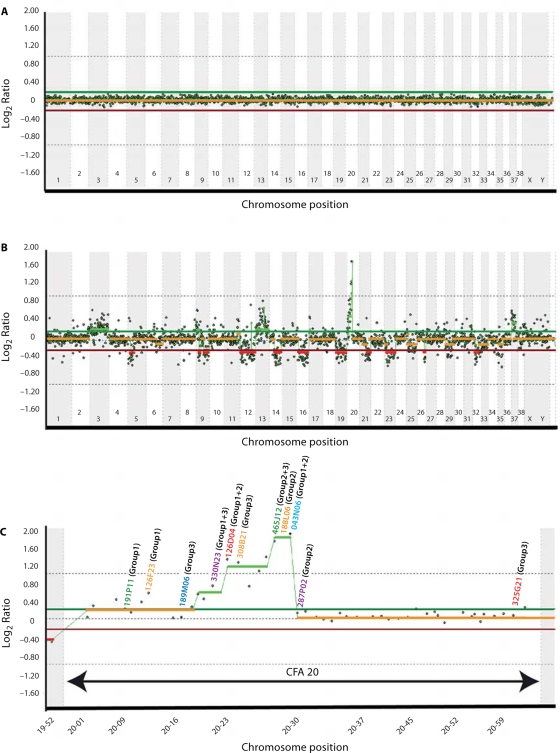
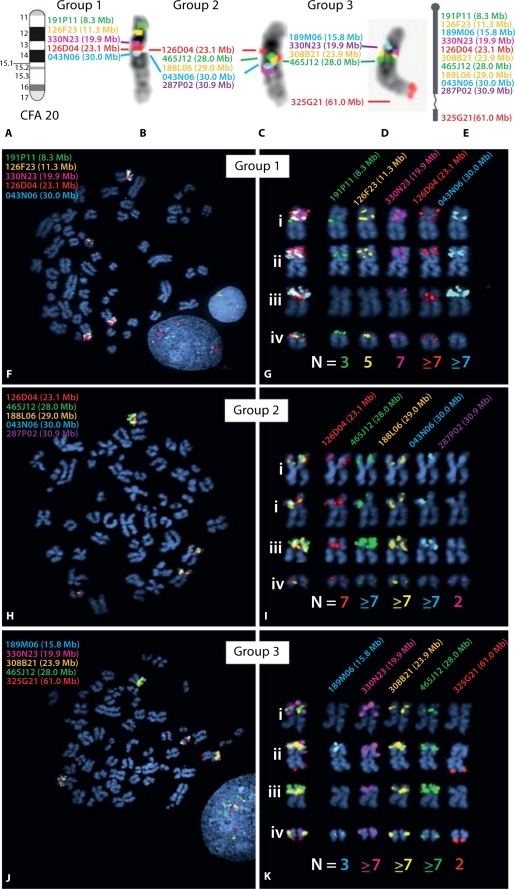
Molecular cytogenetic studies have been instrumental in defining the nature of numerical and structural chromosome changes in human cancers, but their significance remains to be fully understood. The emergence of high quality genome assemblies for several model organisms provides exciting opportunities to develop novel genome-integrated molecular cytogenetic resources that now permit a comparative approach to evaluating the relevance of tumor-associated chromosome aberrations, both within and between species. We have used the dog genome sequence assembly to identify a framework panel of 2,097 bacterial artificial chromosome (BAC) clones, selected at intervals of approximately one megabase. Each clone has been evaluated by multicolor fluorescence in situ hybridization (FISH) to confirm its unique cytogenetic location in concordance with its reported position in the genome assembly, providing new information on the organization of the dog genome. This panel of BAC clones also represents a powerful cytogenetic resource with numerous potential applications. We have used the clone set to develop a genome-wide microarray for comparative genomic hybridization (aCGH) analysis, and demonstrate its application in detection of tumor-associated DNA copy number aberrations (CNAs) including single copy deletions and amplifications, regional aneuploidy and whole chromosome aneuploidy. We also show how individual clones selected from the BAC panel can be used as FISH probes in direct evaluation of tumor karyotypes, to verify and explore CNAs detected using aCGH analysis. This cytogenetically validated, genome integrated BAC clone panel has enormous potential for aiding gene discovery through a comparative approach to molecular oncology.
Copyright 2008 S. Karger AG, Basel.
Figures
References
-
- Backx L, Van Esch H, Melotte C, Kosyakova N, Starke H, et al. Array painting using microdissected chromosomes to map chromosomal breakpoints. Cytogenet Genome Res. 2007;116:158–166. - PubMed
-
- Barrans SL, Fenton JA, Banham A, Owen RG, Jack AS. Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood. 2004;104:2933–2935. - PubMed
-
- Breen M, Modiano JF. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans – man and his best friend share more than companionship. Chromosome Res. 2008;16:145–154. - PubMed
-
- Breen M, Bullerdiek J, Langford CF. The DAPI banded karyotype of the domestic dog (Canis familiaris) generated using chromosome-specific paint probes. Chromosome Res. 1999a;7:401–406. - PubMed
-
- Breen M, Thomas R, Binns MM, Carter NP, Langford CF. Reciprocal chromosome painting reveals detailed regions of conserved synteny between the karyotypes of the domestic dog (Canis familiaris) and human. Genomics. 1999b;61:145–155. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
