

Frequent toggling between alternative amino acids is driven by selection in HIV-1
- PMID: 19096508
- PMCID: PMC2592544
- DOI: 10.1371/journal.ppat.1000242
Frequent toggling between alternative amino acids is driven by selection in HIV-1
Abstract
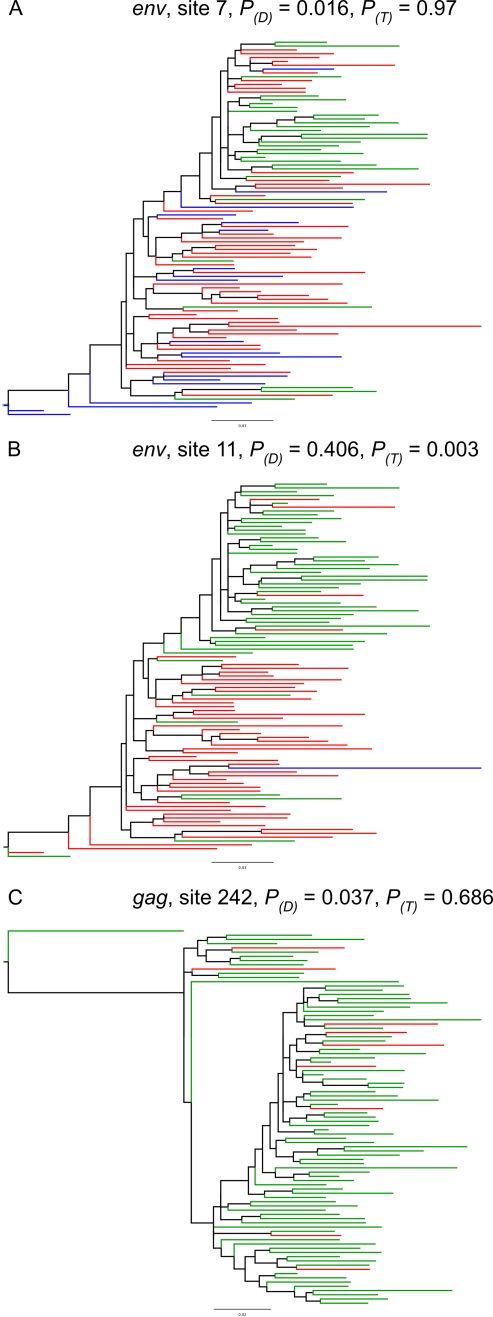
Host immune responses against infectious pathogens exert strong selective pressures favouring the emergence of escape mutations that prevent immune recognition. Escape mutations within or flanking functionally conserved epitopes can occur at a significant cost to the pathogen in terms of its ability to replicate effectively. Such mutations come under selective pressure to revert to the wild type in hosts that do not mount an immune response against the epitope. Amino acid positions exhibiting this pattern of escape and reversion are of interest because they tend to coincide with immune responses that control pathogen replication effectively. We have used a probabilistic model of protein coding sequence evolution to detect sites in HIV-1 exhibiting a pattern of rapid escape and reversion. Our model is designed to detect sites that toggle between a wild type amino acid, which is susceptible to a specific immune response, and amino acids with lower replicative fitness that evade immune recognition. Through simulation, we show that this model has significantly greater power to detect selection involving immune escape and reversion than standard models of diversifying selection, which are sensitive to an overall increased rate of non-synonymous substitution. Applied to alignments of HIV-1 protein coding sequences, the model of immune escape and reversion detects a significantly greater number of adaptively evolving sites in env and nef. In all genes tested, the model provides a significantly better description of adaptively evolving sites than standard models of diversifying selection. Several of the sites detected are corroborated by association between Human Leukocyte Antigen (HLA) and viral sequence polymorphisms. Overall, there is evidence for a large number of sites in HIV-1 evolving under strong selective pressure, but exhibiting low sequence diversity. A phylogenetic model designed to detect rapid toggling between wild type and escape amino acids identifies a larger number of adaptively evolving sites in HIV-1, and can in some cases correctly identify the amino acid that is susceptible to the immune response.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

 , where c represents the codon class. The
, where c represents the codon class. The  parameters take account of the codon bias estimated across the entire alignment and free parameters tc which describe the proportion of time spent by the site in each of the three codon classes.
parameters take account of the codon bias estimated across the entire alignment and free parameters tc which describe the proportion of time spent by the site in each of the three codon classes.


References
-
- Goulder PJR, Watkins DI. HIV and SIV CTL escape: Implications for vaccine design. Nature Reviews Immunology. 2004;4:630–640. - PubMed
-
- Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–289. - PubMed
-
- Serwold T, Shastri N. Specific proteolytic cleavages limit the diversity of the pool of peptides available to MHC class I molecules in living cells. J Immunol. 1999;162:4712–4719. - PubMed
-
- Brumme ZL, Tao I, Szeto S, Brumme CJ, Carlson JM, et al. Human leukocyte antigen-specific polymorphisms in HIV-1 Gag and their association with viral load in chronic untreated infection. Aids. 2008;22:1277–1286. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
