

Induced pluripotent stem cells from a spinal muscular atrophy patient
- PMID: 19098894
- PMCID: PMC2659408
- DOI: 10.1038/nature07677
Induced pluripotent stem cells from a spinal muscular atrophy patient
Abstract
Spinal muscular atrophy is one of the most common inherited forms of neurological disease leading to infant mortality. Patients have selective loss of lower motor neurons resulting in muscle weakness, paralysis and often death. Although patient fibroblasts have been used extensively to study spinal muscular atrophy, motor neurons have a unique anatomy and physiology which may underlie their vulnerability to the disease process. Here we report the generation of induced pluripotent stem cells from skin fibroblast samples taken from a child with spinal muscular atrophy. These cells expanded robustly in culture, maintained the disease genotype and generated motor neurons that showed selective deficits compared to those derived from the child's unaffected mother. This is the first study to show that human induced pluripotent stem cells can be used to model the specific pathology seen in a genetically inherited disease. As such, it represents a promising resource to study disease mechanisms, screen new drug compounds and develop new therapies.
Figures
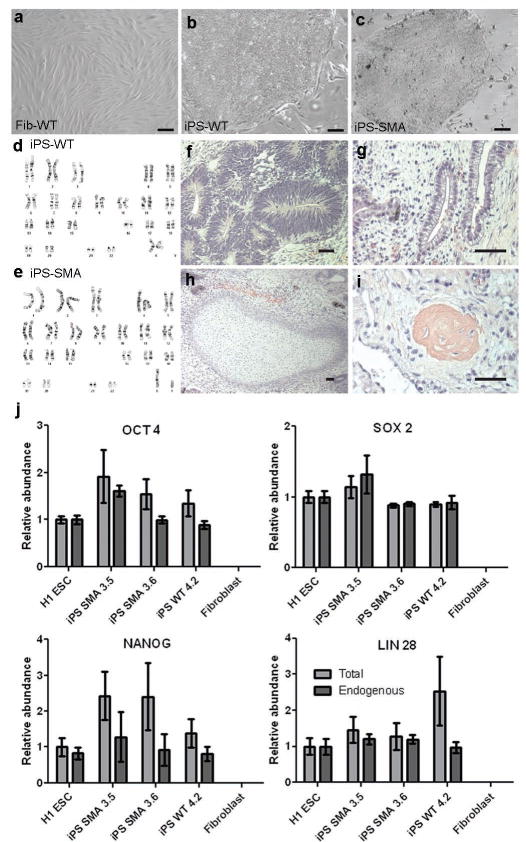
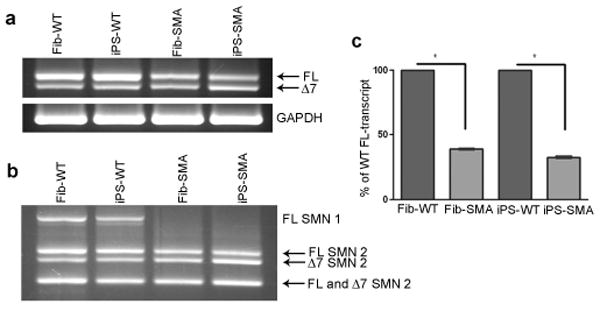
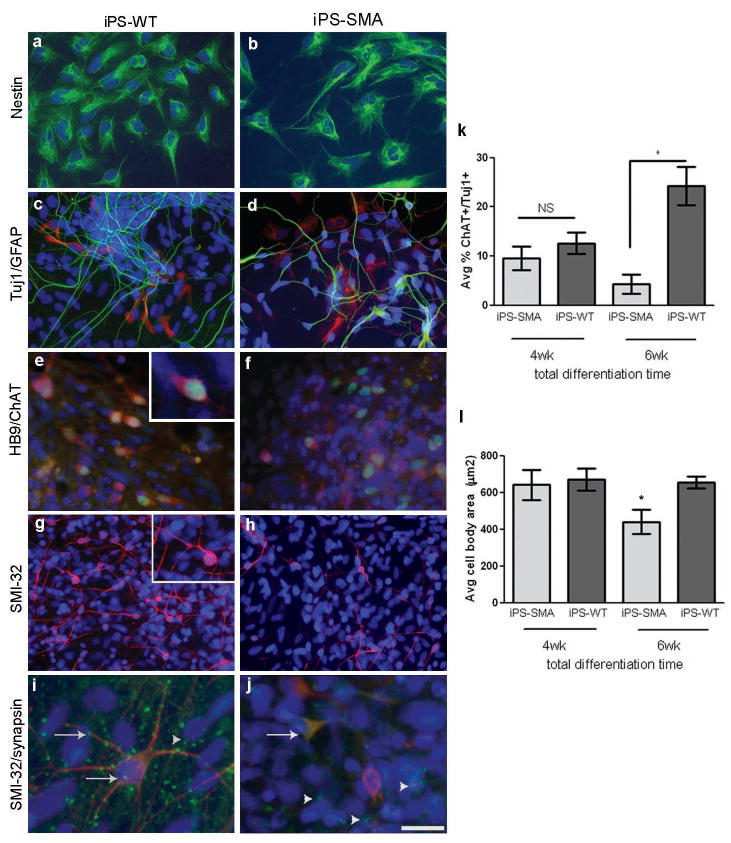
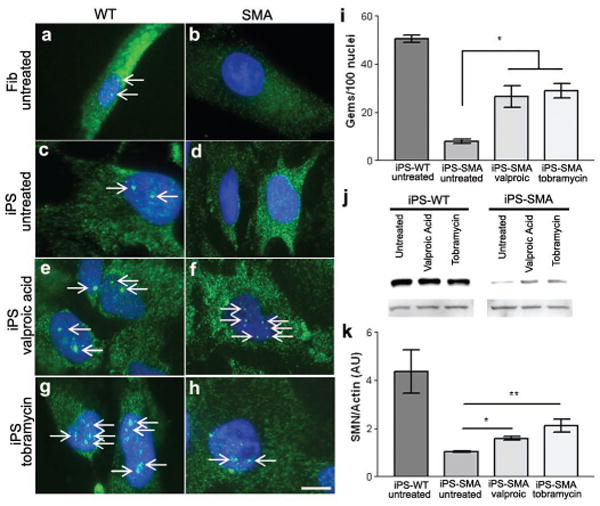
Comment in
-
Stem cells: Tailor-made diseased neurons.Nature. 2009 Jan 15;457(7227):269-70. doi: 10.1038/457269a. Nature. 2009. PMID: 19148087 No abstract available.
References
-
- Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Coovert DD, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. - PubMed
-
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. - PubMed
-
- Munsat TL, Davies KE. Neuromuscul Disord; International SMA consortium meeting; 26-28 June 1992; Bonn, Germany. 1992. pp. 423–428. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
