

Accelerated genetic drift on chromosome X during the human dispersal out of Africa
- PMID: 19098910
- PMCID: PMC2612098
- DOI: 10.1038/ng.303
Accelerated genetic drift on chromosome X during the human dispersal out of Africa
Abstract
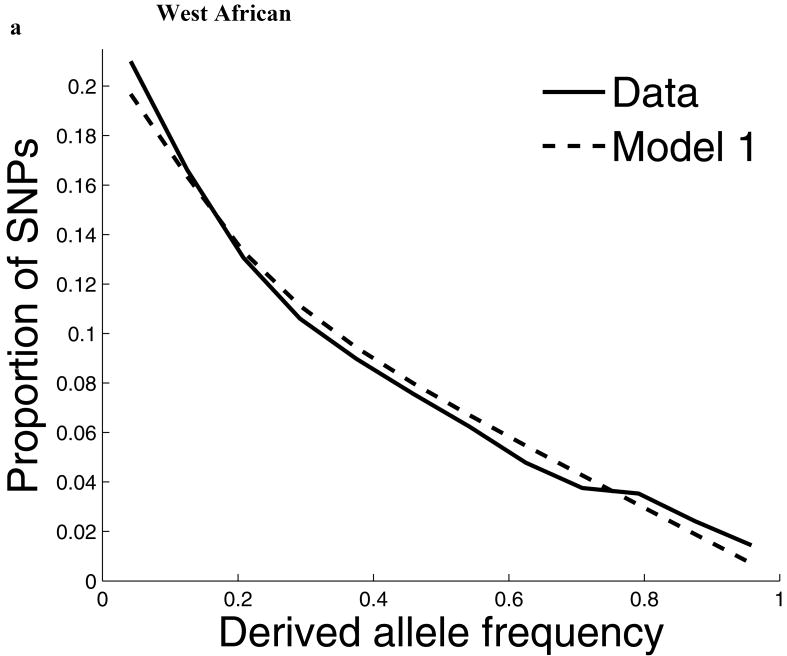
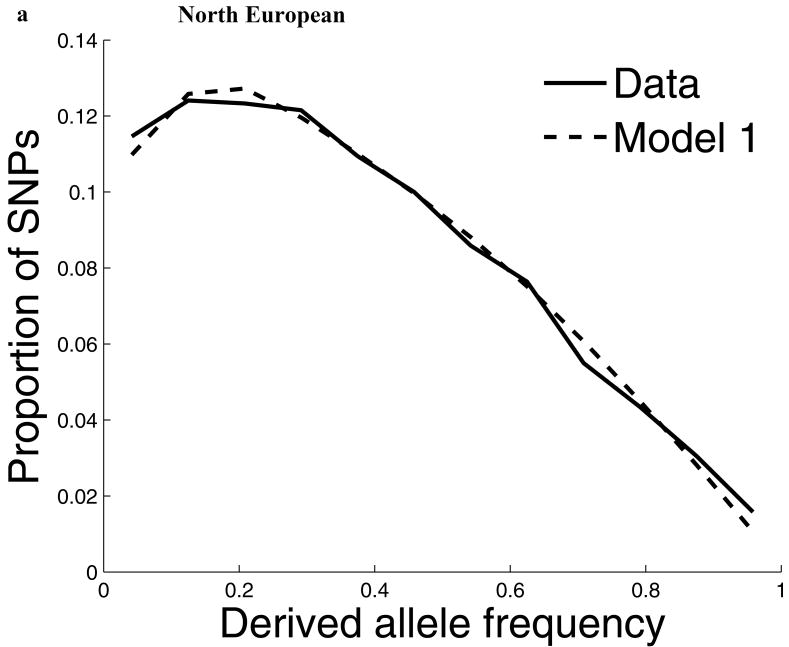
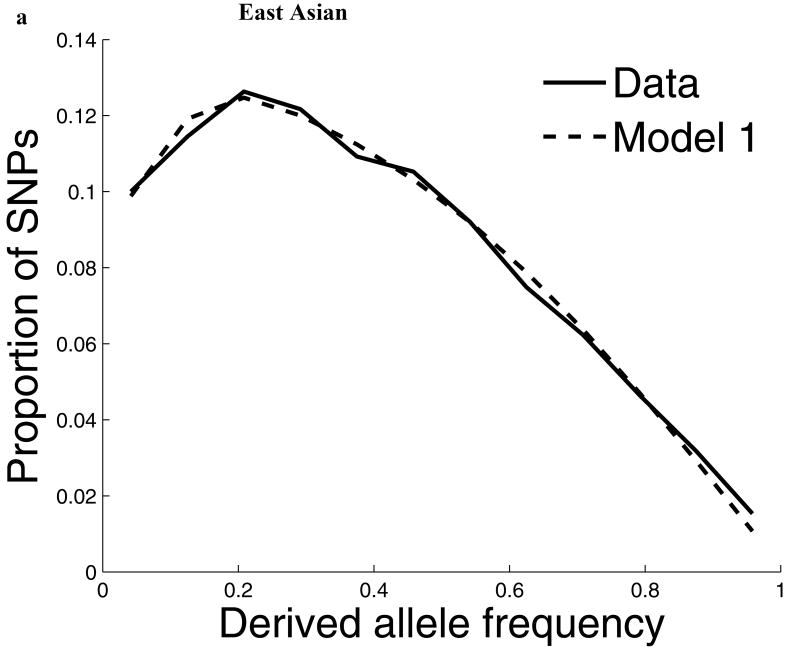
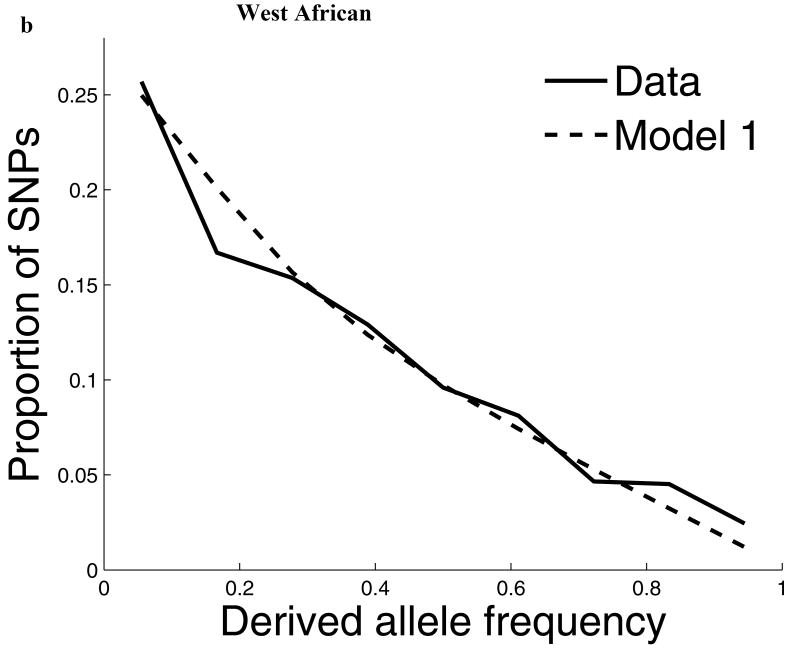
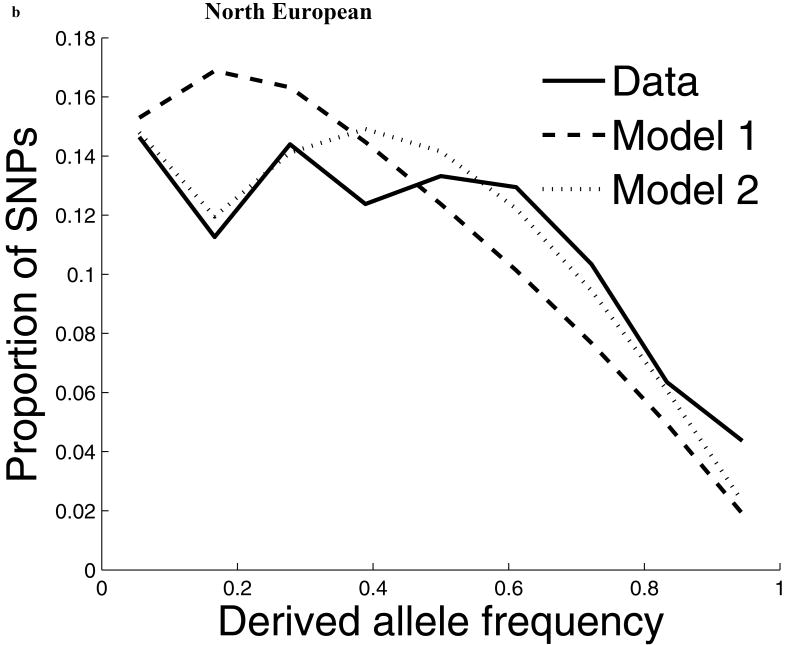
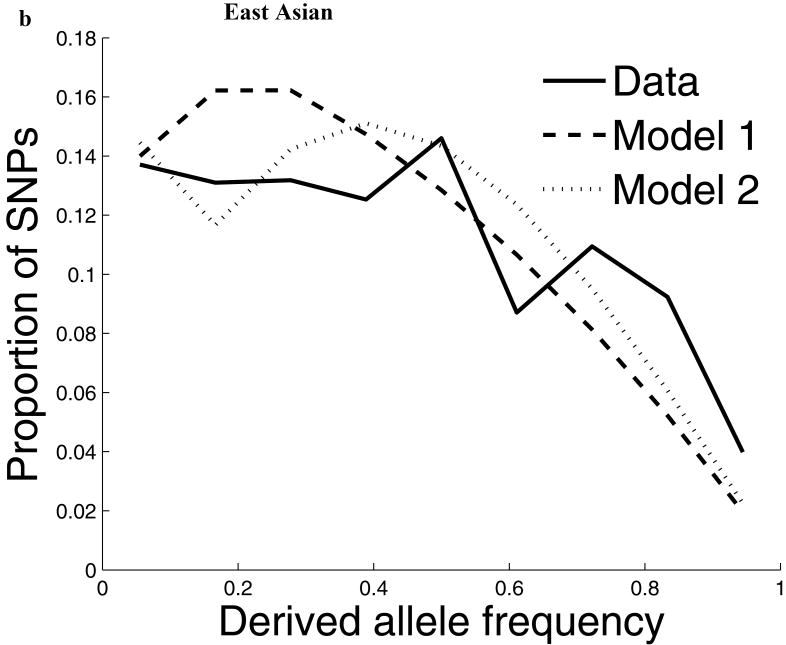
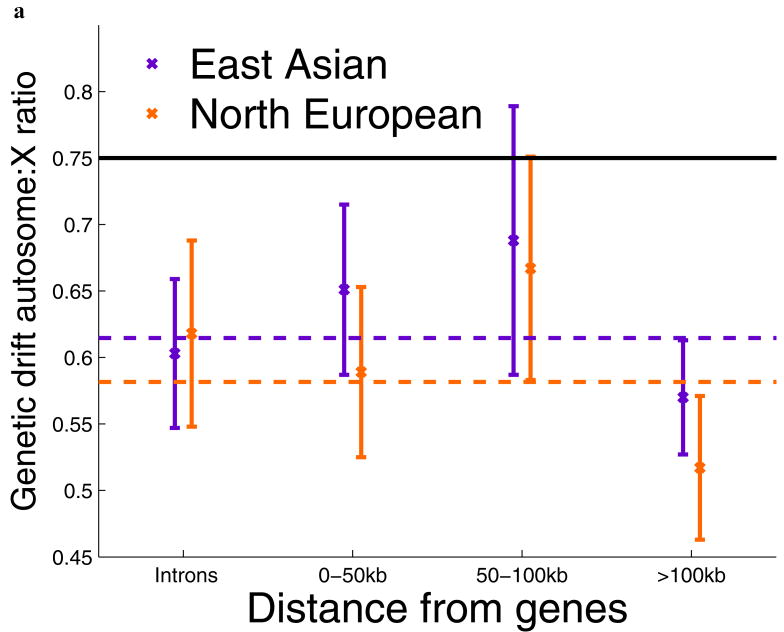
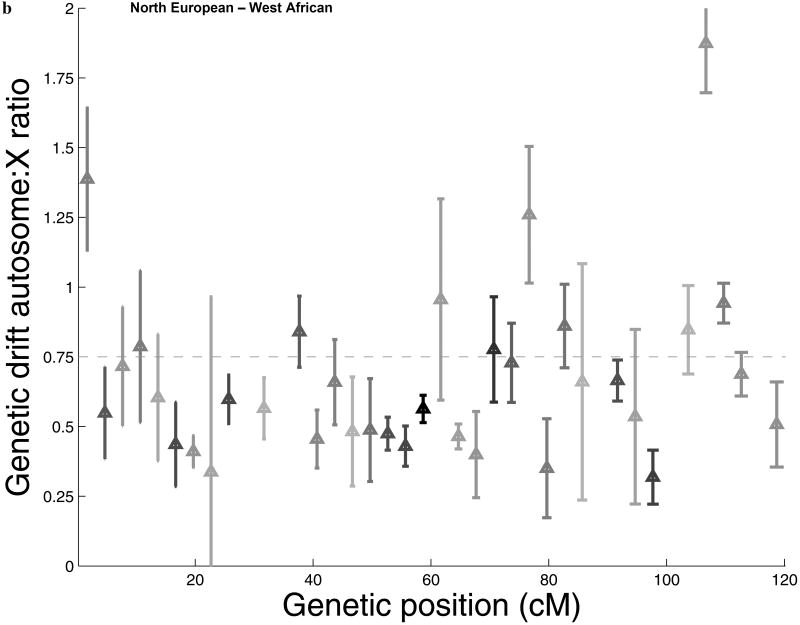
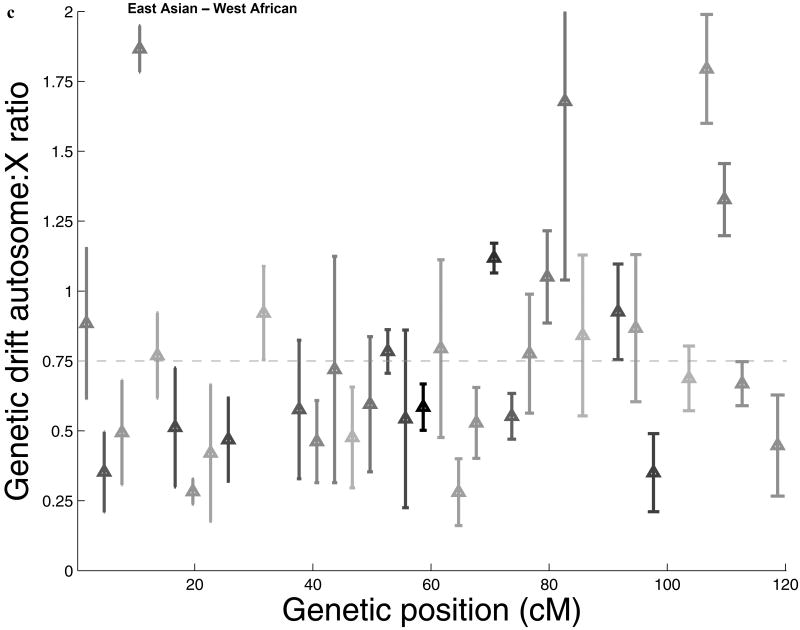
Comparisons of chromosome X and the autosomes can illuminate differences in the histories of males and females as well as shed light on the forces of natural selection. We compared the patterns of variation in these parts of the genome using two datasets that we assembled for this study that are both genomic in scale. Three independent analyses show that around the time of the dispersal of modern humans out of Africa, chromosome X experienced much more genetic drift than is expected from the pattern on the autosomes. This is not predicted by known episodes of demographic history, and we found no similar patterns associated with the dispersals into East Asia and Europe. We conclude that a sex-biased process that reduced the female effective population size, or an episode of natural selection unusually affecting chromosome X, was associated with the founding of non-African populations.
Figures
Comment in
-
Evaluating signatures of sex-specific processes in the human genome.Nat Genet. 2009 Jan;41(1):8-10. doi: 10.1038/ng0109-8. Nat Genet. 2009. PMID: 19112457 Free PMC article.
References
-
- Li JZ, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–4. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
