

Unscheduled Akt-triggered activation of cyclin-dependent kinase 2 as a key effector mechanism of apoptin's anticancer toxicity
- PMID: 19103742
- PMCID: PMC2643822
- DOI: 10.1128/MCB.00668-08
Unscheduled Akt-triggered activation of cyclin-dependent kinase 2 as a key effector mechanism of apoptin's anticancer toxicity
Abstract
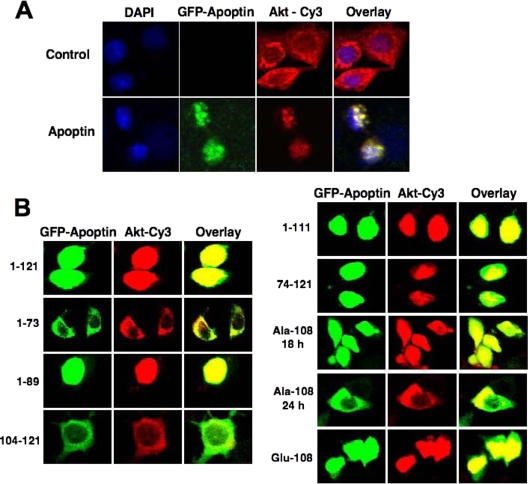
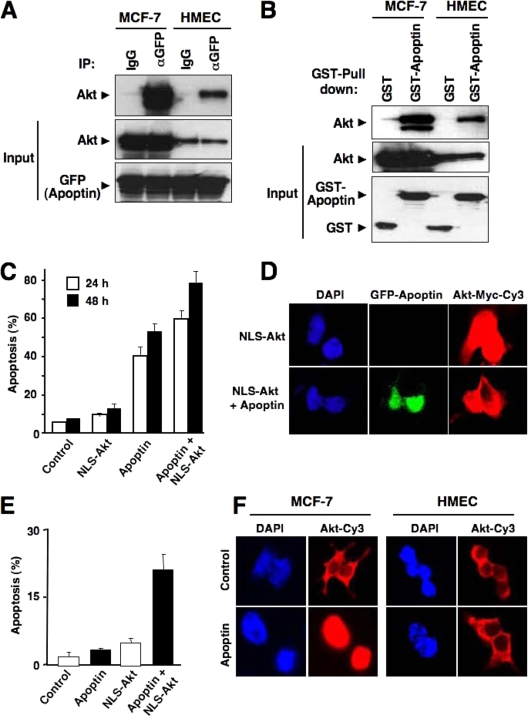
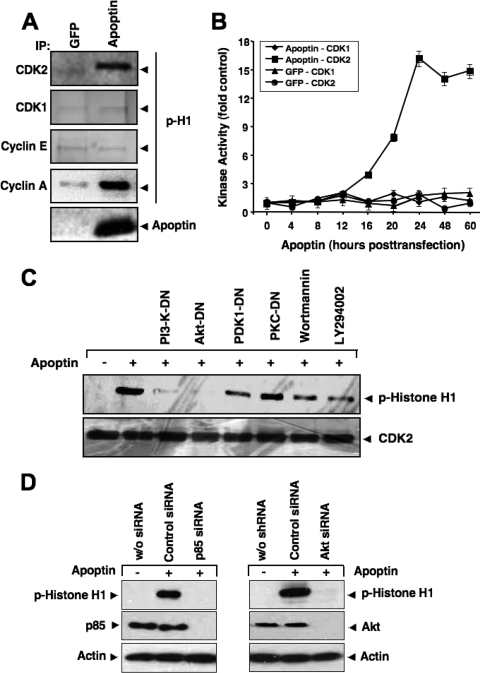
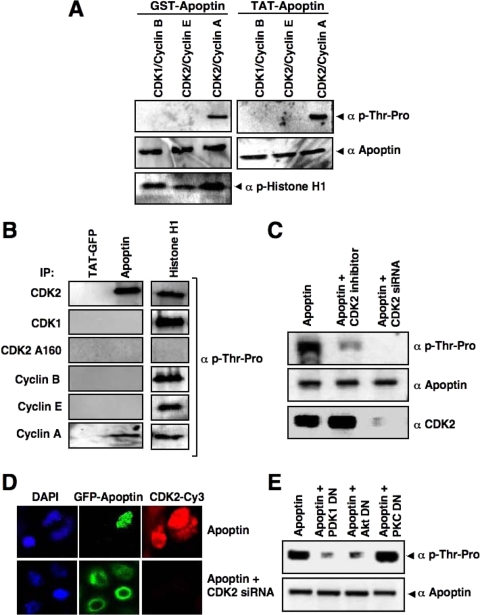
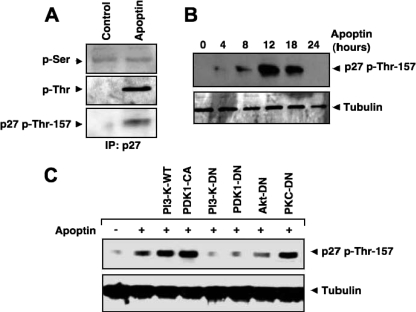
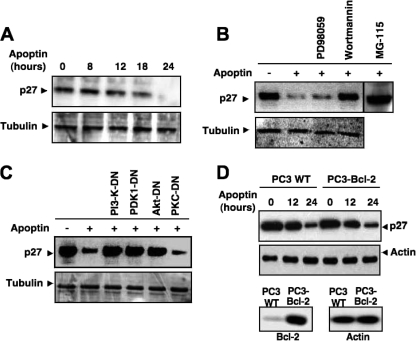
Apoptin, a protein from the chicken anemia virus, has attracted attention because it specifically kills tumor cells while leaving normal cells unharmed. The reason for this tumor selectivity is unclear and depends on subcellular localization, as apoptin resides in the cytoplasm of normal cells but in the nuclei of transformed cells. It was shown that nuclear localization and tumor-specific killing crucially require apoptin's phosphorylation by an as yet unknown kinase. Here we elucidate the pathway of apoptin-induced apoptosis and show that it essentially depends on abnormal phosphatidylinositol 3-kinase (PI3-kinase)/Akt activation, resulting in the activation of the cyclin-dependent kinase CDK2. Inhibitors as well as dominant-negative mutants of PI3-kinase and Akt not only inhibited CDK2 activation but also protected cells from apoptin-induced cell death. Akt activated CDK2 by direct phosphorylation as well as by the phosphorylation-induced degradation of the inhibitor p27(Kip1). Importantly, we also identified CDK2 as the principal kinase that phosphorylates apoptin and is crucially required for apoptin-induced cell death. Immortalized CDK2-deficient fibroblasts and CDK2 knockdown cells were markedly protected against apoptin. Thus, our results not only decipher the pathway of apoptin-induced cell death but also provide mechanistic insights for the selective killing of tumor cells.
Figures



Similar articles
-
Activation of the Chicken Anemia Virus Apoptin Protein by Chk1/2 Phosphorylation Is Required for Apoptotic Activity and Efficient Viral Replication.J Virol. 2016 Sep 29;90(20):9433-45. doi: 10.1128/JVI.00936-16. Print 2016 Oct 15. J Virol. 2016. PMID: 27512067 Free PMC article.
-
Apoptin, a tumor-selective killer.Biochim Biophys Acta. 2009 Aug;1793(8):1335-42. doi: 10.1016/j.bbamcr.2009.04.002. Epub 2009 Apr 15. Biochim Biophys Acta. 2009. PMID: 19374922 Review.
-
Mechanisms of Apoptin-induced cell death.Med Oncol. 2012 Dec;29(4):2985-91. doi: 10.1007/s12032-011-0119-2. Epub 2011 Nov 22. Med Oncol. 2012. PMID: 22105148 Review.
-
The viral death effector Apoptin reveals tumor-specific processes.Apoptosis. 2004 May;9(3):315-22. doi: 10.1023/b:appt.0000025808.48885.9c. Apoptosis. 2004. PMID: 15258463 Review.
-
Development and application of an in vitro apoptin kinase assay.Anal Biochem. 2012 Feb 1;421(1):68-74. doi: 10.1016/j.ab.2011.10.030. Epub 2011 Oct 22. Anal Biochem. 2012. PMID: 22080040
Cited by
-
Nuclear localized Akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1).Cell Cycle. 2015;14(13):2109-20. doi: 10.1080/15384101.2015.1041692. Epub 2015 Jun 1. Cell Cycle. 2015. PMID: 26030190 Free PMC article.
-
IRF8 induces senescence of lung cancer cells to exert its tumor suppressive function.Cell Cycle. 2019 Dec;18(23):3300-3312. doi: 10.1080/15384101.2019.1674053. Epub 2019 Oct 9. Cell Cycle. 2019. PMID: 31594449 Free PMC article.
-
Cyclin-dependent kinase activity controls the onset of the HCMV lytic cycle.PLoS Pathog. 2010 Sep 9;6(9):e1001096. doi: 10.1371/journal.ppat.1001096. PLoS Pathog. 2010. PMID: 20844576 Free PMC article.
-
Activation of the Chicken Anemia Virus Apoptin Protein by Chk1/2 Phosphorylation Is Required for Apoptotic Activity and Efficient Viral Replication.J Virol. 2016 Sep 29;90(20):9433-45. doi: 10.1128/JVI.00936-16. Print 2016 Oct 15. J Virol. 2016. PMID: 27512067 Free PMC article.
-
Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells.Chem Res Toxicol. 2010 Mar 15;23(3):568-77. doi: 10.1021/tx9003193. Chem Res Toxicol. 2010. PMID: 20112989 Free PMC article.
References
-
- Aki, T., K. Yamaguchi, T. Fujimiya, and Y. Mizukami. 2003. Phosphoinositide 3-kinase accelerates autophagic cell death during glucose deprivation in the rat cardiomyocyte-derived cell line H9c2. Oncogene 228529-8535. - PubMed
-
- Backendorf, C., A. E. Visser, A. G. de Boer, R. Zimmerman, M. Visser, P. Voskamp, Y. H. Zhang, and M. H. Noteborn. 2008. Apoptin: therapeutic potential of an early sensor of carcinogenic transformation. Annu. Rev. Pharmacol. Toxicol. 48143-169. - PubMed
-
- Barkett, M., and T. D. Gilmore 1999. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 186910-6924. - PubMed
-
- Bashir, T., N. V. Dorrello, V. Amador, D. Guardavaccaro, and M. Pagano. 2004. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428190-193. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous