

Comparing platforms for C. elegans mutant identification using high-throughput whole-genome sequencing
- PMID: 19107202
- PMCID: PMC2603312
- DOI: 10.1371/journal.pone.0004012
Comparing platforms for C. elegans mutant identification using high-throughput whole-genome sequencing
Abstract
Background: Whole-genome sequencing represents a promising approach to pinpoint chemically induced mutations in genetic model organisms, thereby short-cutting time-consuming genetic mapping efforts.
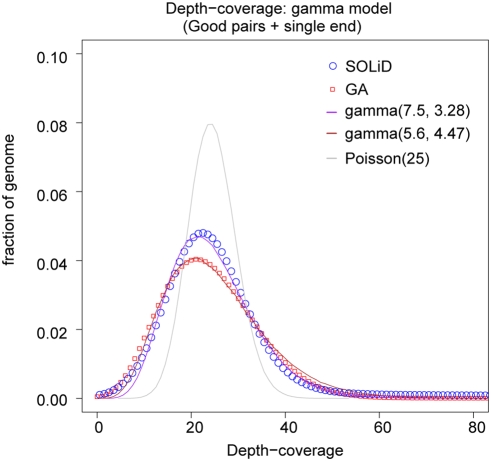
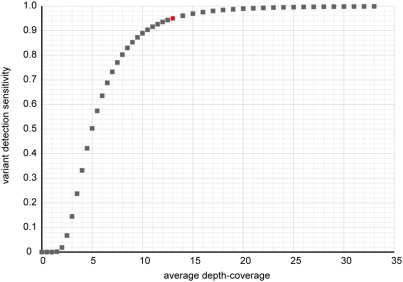
Principal findings: We compare here the ability of two leading high-throughput platforms for paired-end deep sequencing, SOLiD (ABI) and Genome Analyzer (Illumina; "Solexa"), to achieve the goal of mutant detection. As a test case we used a mutant C. elegans strain that harbors a mutation in the lsy-12 locus which we compare to the reference wild-type genome sequence. We analyzed the accuracy, sensitivity, and depth-coverage characteristics of the two platforms. Both platforms were able to identify the mutation that causes the phenotype of the mutant C. elegans strain, lsy-12. Based on a 4 MB genomic region in which individual variants were validated by Sanger sequencing, we observe tradeoffs between rates of false positives and false negatives when using both platforms under similar coverage and mapping criteria.
Significance: In conclusion, whole-genome sequencing conducted by either platform is a viable approach for the identification of single-nucleotide variations in the C. elegans genome.
Conflict of interest statement
Figures
References
-
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
