

Glucose and NADPH oxidase drive neuronal superoxide formation in stroke
- PMID: 19107988
- PMCID: PMC4304737
- DOI: 10.1002/ana.21511
Glucose and NADPH oxidase drive neuronal superoxide formation in stroke
Abstract
Objective: Hyperglycemia has been recognized for decades to be an exacerbating factor in ischemic stroke, but the mechanism of this effect remains unresolved. Here, we evaluated superoxide production by neuronal nicotinamide adenine dinucleotide phosphate (NADPH) oxidase as a possible link between glucose metabolism and neuronal death in ischemia-reperfusion.
Methods: Superoxide production was measured by the ethidium method in cultured neurons treated with oxygen-glucose deprivation and in mice treated with forebrain ischemia-reperfusion. The role of NADPH oxidase was examined using genetic disruption of its p47(phox) subunit and with the pharmacological inhibitor apocynin.
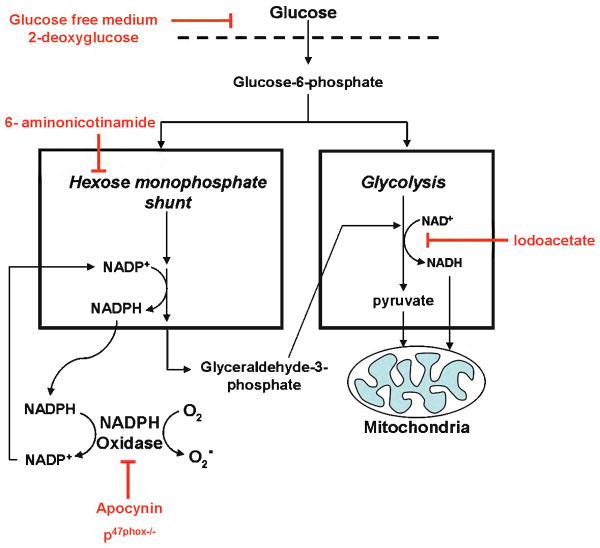
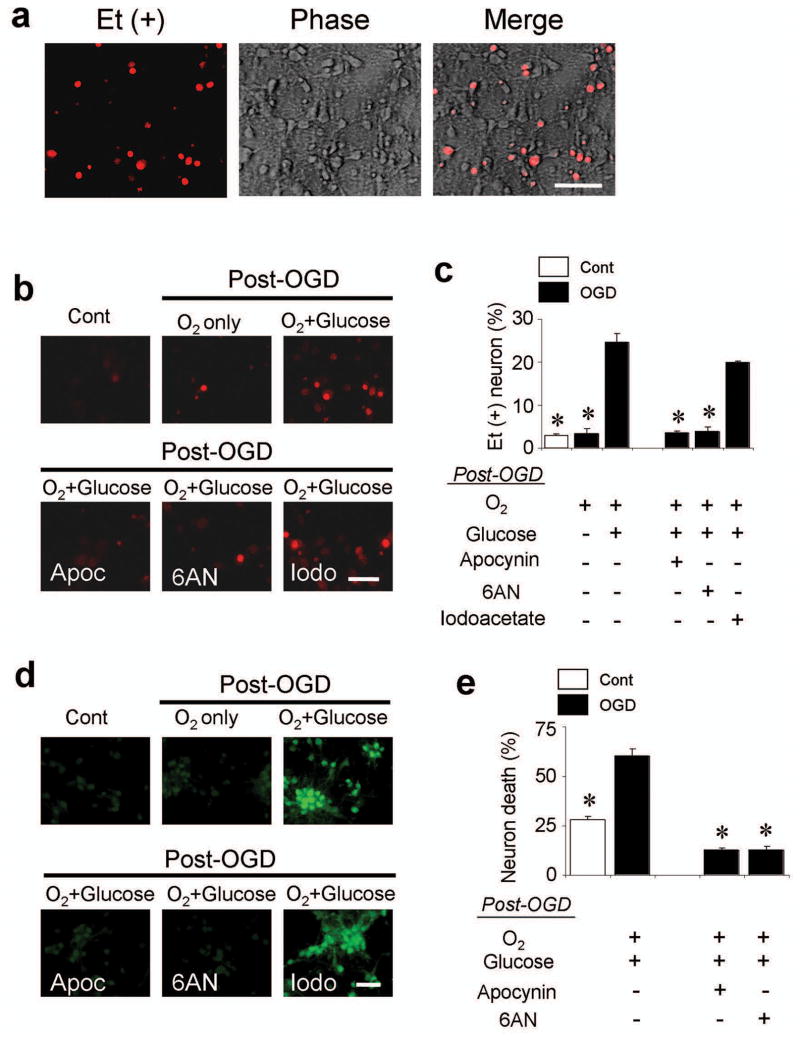
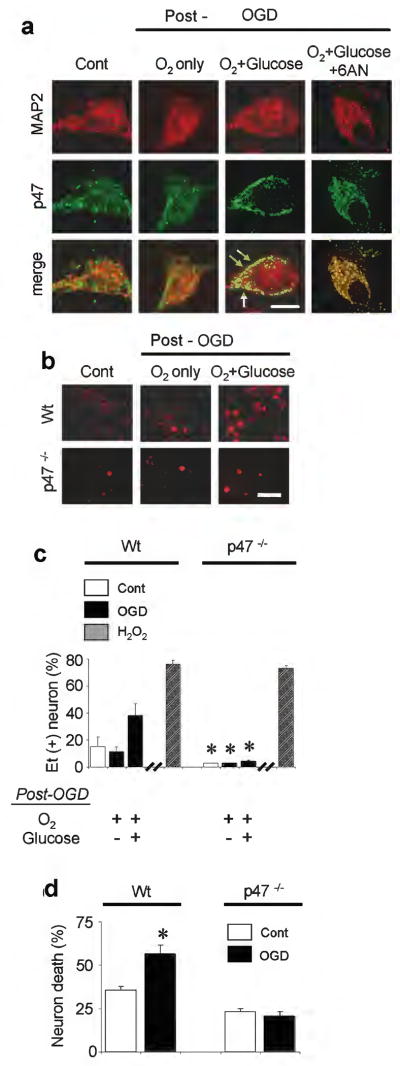
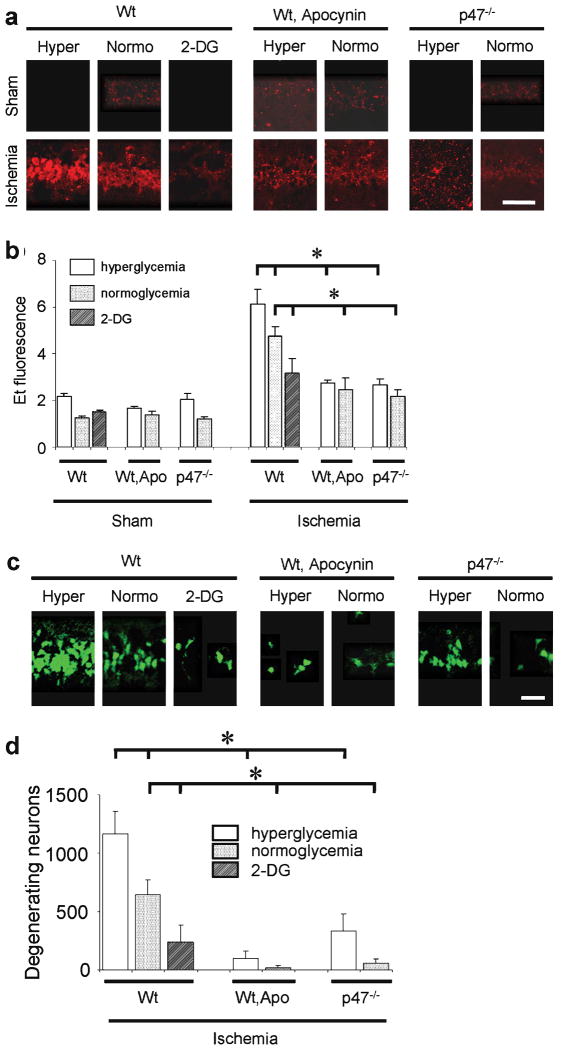
Results: In neuron cultures, postischemic superoxide production and cell death were completely prevented by removing glucose from the medium, by inactivating NADPH oxidase, or by inhibiting the hexose monophosphate shunt that generates NADPH from glucose. In murine stroke, neuronal superoxide production and death were decreased by the glucose antimetabolite 2-deoxyglucose and increased by high blood glucose concentrations. Inactivating NADPH oxidase with either apocynin or deletion of the p47(phox) subunit blocked neuronal superoxide production and negated the deleterious effects of hyperglycemia.
Interpretation: These findings identify glucose as the requisite electron donor for reperfusion-induced neuronal superoxide production and establish a previously unrecognized mechanism by which hyperglycemia can exacerbate ischemic brain injury.
Conflict of interest statement
Conflict of interest statement: None of the authors have any financial interest relevant to the work presented in this manuscript.
Figures
References
-
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. N Engl J Med. 1995;333:1581–1587. - PubMed
-
- Zivin JA, Lyden PD, DeGirolami U, et al. Tissue plasminogen activator. Reduction of neurologic damage after experimental embolic stroke. Arch Neurol. 1988;45:387–391. - PubMed
-
- Aronowski J, Strong R, Grotta JC. Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1048–1056. - PubMed
-
- Hallenbeck JM, Dutka AJ. Background review and current concepts of reperfusion injury. Arch Neurol. 1990;47:1245–1254. - PubMed
-
- Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab. 2007;27:435–451. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
