

Nociceptors are interleukin-1beta sensors
- PMID: 19109489
- PMCID: PMC2690713
- DOI: 10.1523/JNEUROSCI.3795-08.2008
Nociceptors are interleukin-1beta sensors
Abstract
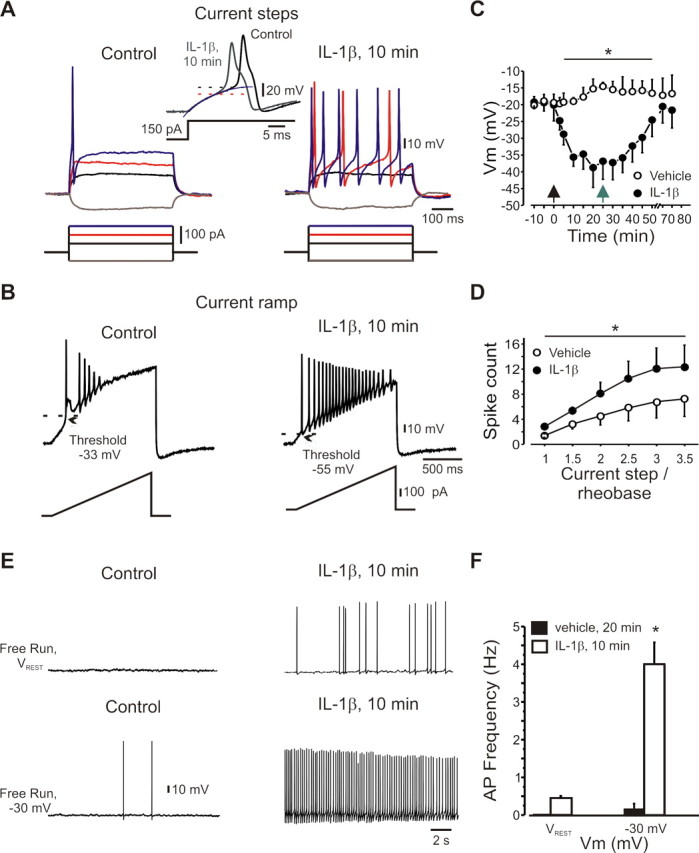
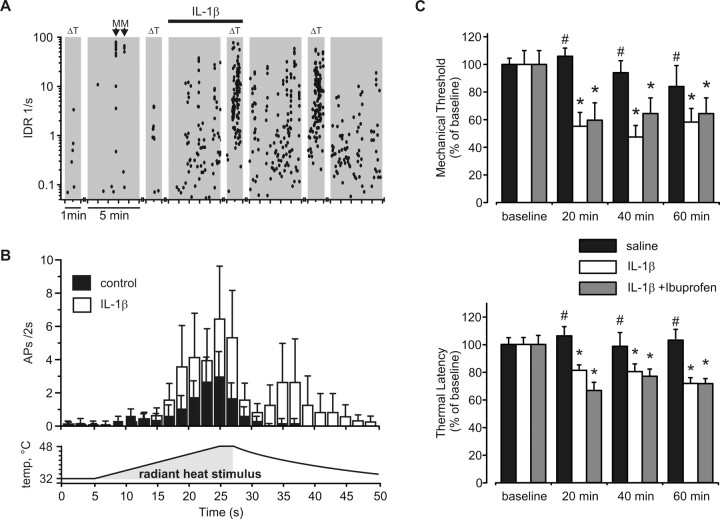
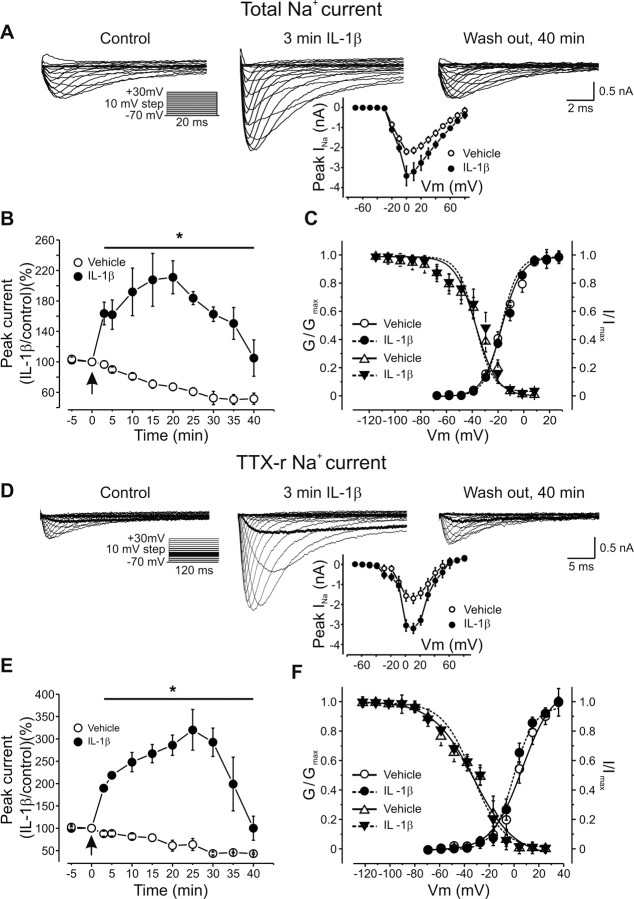
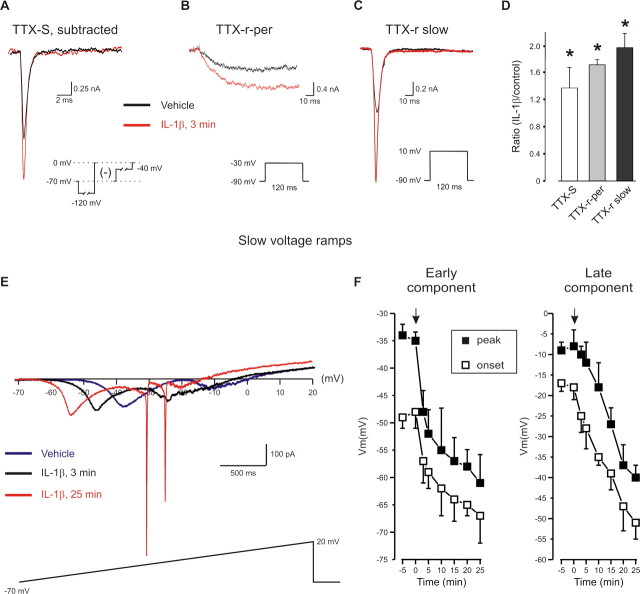
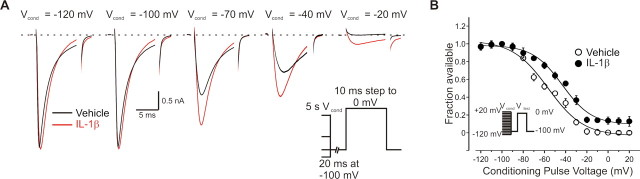
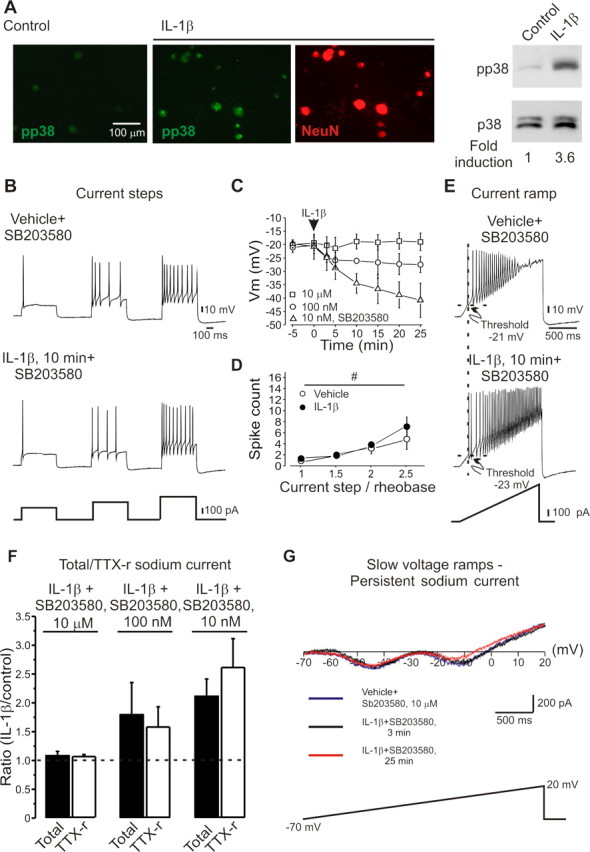
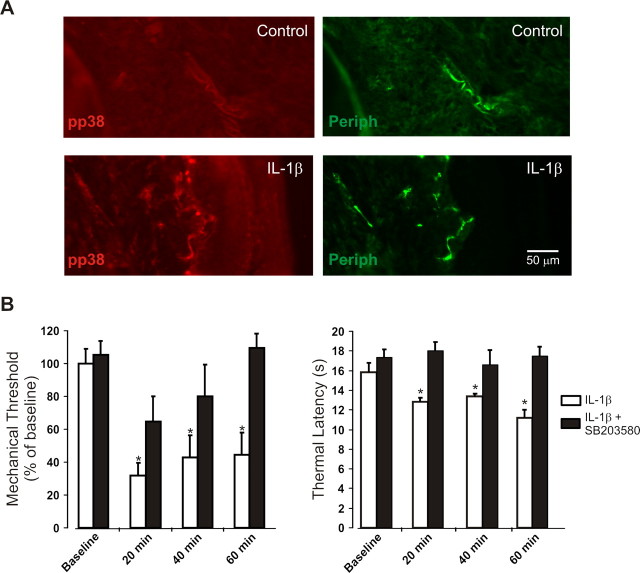
A cardinal feature of inflammation is heightened pain sensitivity at the site of the inflamed tissue. This results from the local release by immune and injured cells of nociceptor sensitizers, including prostaglandin E(2), bradykinin, and nerve growth factor, that reduce the threshold and increase the excitability of the peripheral terminals of nociceptors so that they now respond to innocuous stimuli: the phenomenon of peripheral sensitization. We show here that the proinflammatory cytokine interleukin-1beta (IL-1beta), in addition to producing inflammation and inducing synthesis of several nociceptor sensitizers, also rapidly and directly activates nociceptors to generate action potentials and induce pain hypersensitivity. IL-1beta acts in a p38 mitogen-activated protein kinase (p38 MAP kinase)-dependent manner, to increase the excitability of nociceptors by relieving resting slow inactivation of tetrodotoxin-resistant voltage-gated sodium channels and also enhances persistent TTX-resistant current near threshold. By acting as an IL-1beta sensor, nociceptors can directly signal the presence of ongoing tissue inflammation.
Figures
References
-
- Abdulla FA, Smith PA. Axotomy- and autotomy-induced changes in the excitability of rat dorsal root ganglion neurons. J Neurophysiol. 2001;85:630–643. - PubMed
-
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. - PubMed
-
- Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, Chessell IP, Tate S, Green PJ, Woolf CJ. The voltage-gated sodium channel Na(v) 1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852–12860. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical