

Secondary lipid accumulation in lysosomal disease
- PMID: 19111580
- PMCID: PMC4382014
- DOI: 10.1016/j.bbamcr.2008.11.014
Secondary lipid accumulation in lysosomal disease
Abstract
Lysosomal diseases are inherited metabolic disorders caused by defects in a wide spectrum of lysosomal and a few non-lysosomal proteins. In most cases a single type of primary storage material is identified, which has been used to name and classify the disorders: hence the terms sphingolipidoses, gangliosidoses, mucopolysaccharidoses, glycoproteinoses, and so forth. In addition to this primary storage, however, a host of secondary storage products can also be identified, more often than not having no direct link to the primary protein defect. Lipids - glycosphingolipids and phospholipids, as well as cholesterol - are the most ubiquitous and best studied of these secondary storage materials. While in the past typically considered nonspecific and nonconsequential features of these diseases, newer studies suggest direct links between secondary storage and disease pathogenesis and support the view that understanding all aspects of this sequestration process will provide important insights into the cell biology and treatment of lysosomal disease.
Figures

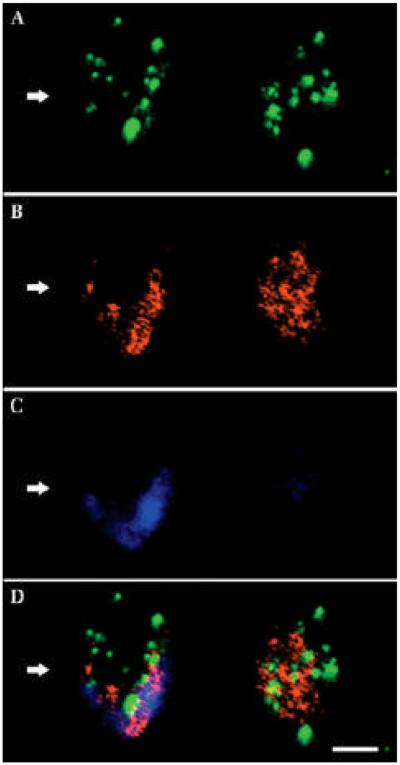
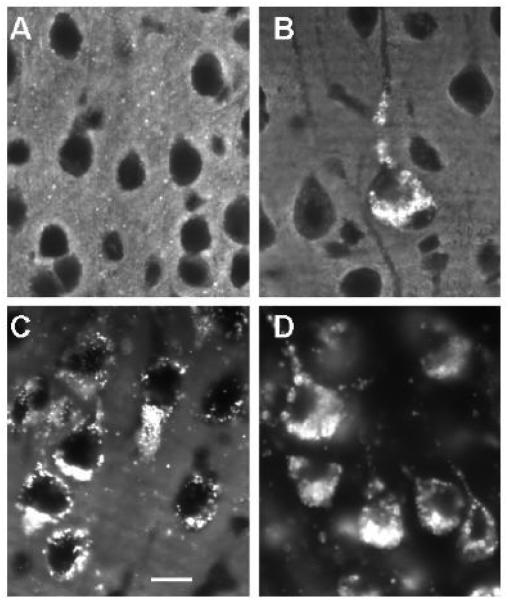
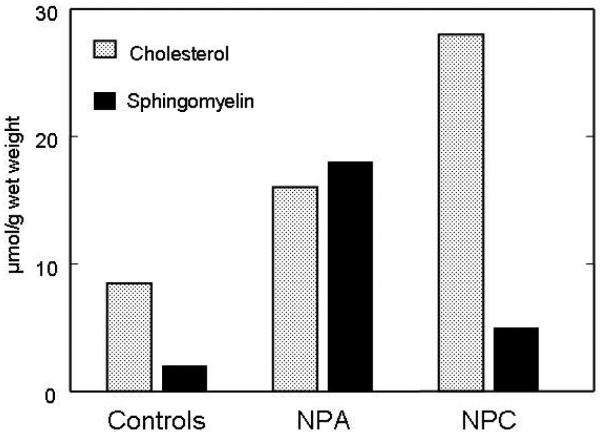
References
-
- Platt FM, Walkley SU. Lysosomal Defects and Storage. In: Platt FM, Walkley SU, editors. Lysosomal Disorders of the Brain. Oxford University Press; Oxford: 2004. pp. 32–49.
-
- Cumings JN. Abnormalities in lipid metabolism in two members of a amily with Niemann-Pick disease. In: Aronson SM, Volk BW, editors. Cerebral lipidoses. Academic Press; New York: 1962. pp. 171–178.
-
- Kamoshita S, Aron AM, Suzuki K, Suzuki K. Infantile Niemann-Pick disease. A chemical study with isolation and characterization of membranous cytoplasmic bodies and myelin. Am. J. Dis. Child. 1969;117:379–394. - PubMed
-
- Martin JJ, Philippart M, Van Hauwaert J, Callahan JW, Deberdt R. Niemann-Pick disease (Crocker's group A). Late onset and pigmentary degeneration resembling Hallervorden-Spatz syndrome. Arch. Neurol. 1972;27:45–51. - PubMed
-
- Brunngraber EG, Berra B, Zambotti V. Altered levels of tissue glycoproteins, gangliosides, glycosaminoglycans and lipids in Niemann-Pick's disease. Clin. Chim. Acta. 1973;48:173–181. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
