

The skeletal L-type Ca(2+) current is a major contributor to excitation-coupled Ca(2+) entry
- PMID: 19114636
- PMCID: PMC2606935
- DOI: 10.1085/jgp.200810105
The skeletal L-type Ca(2+) current is a major contributor to excitation-coupled Ca(2+) entry
Abstract
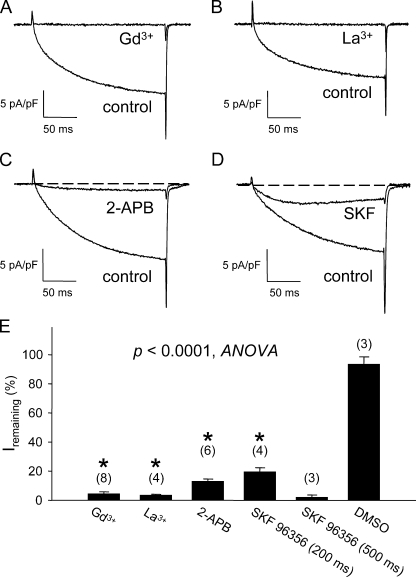
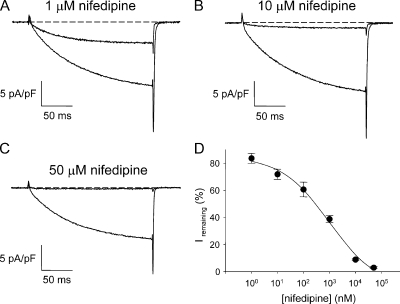
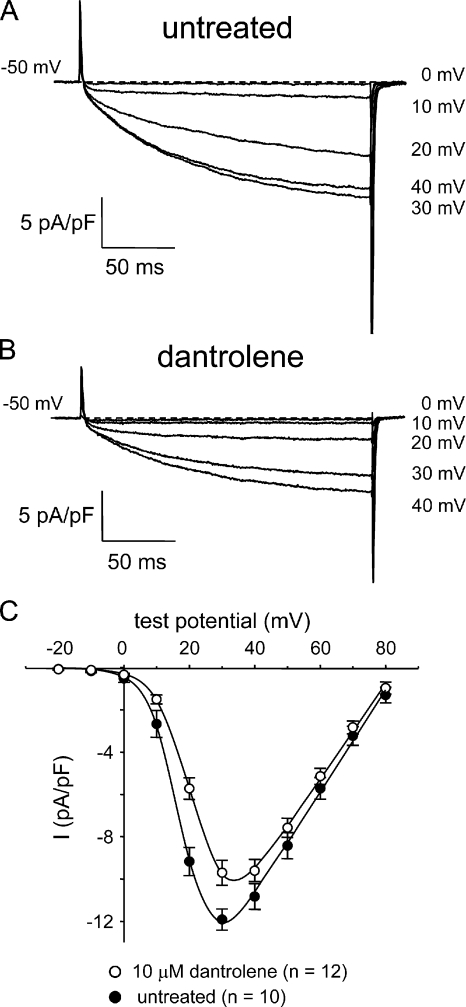
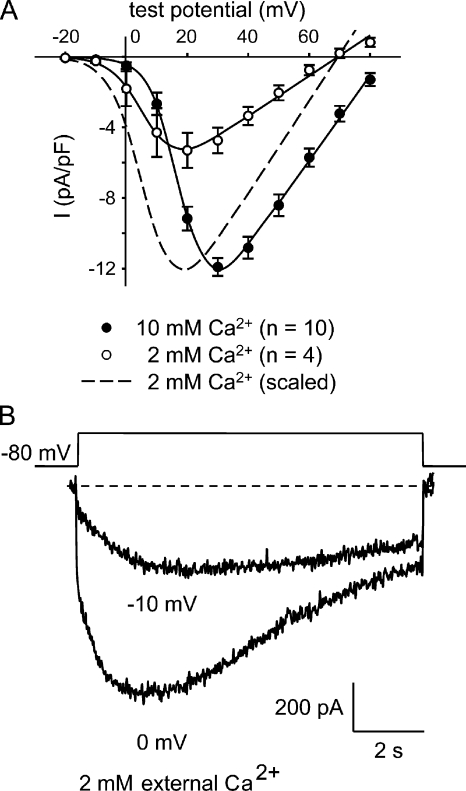
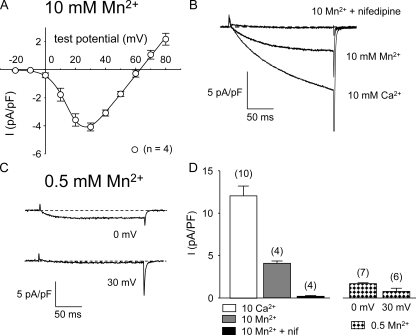
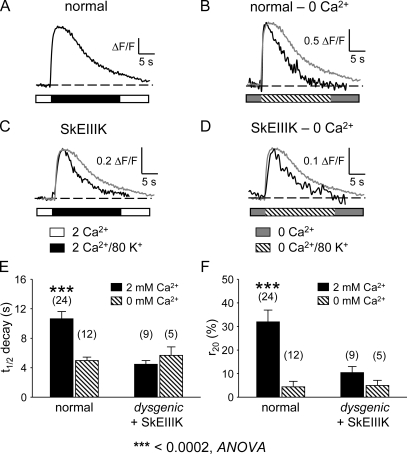
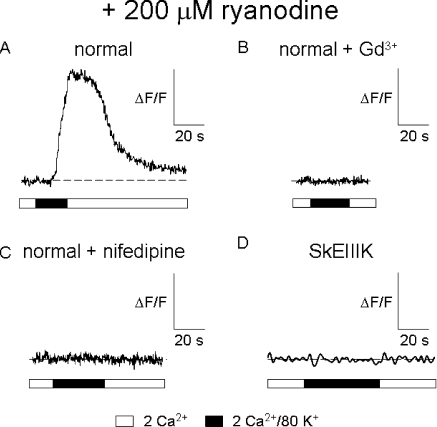
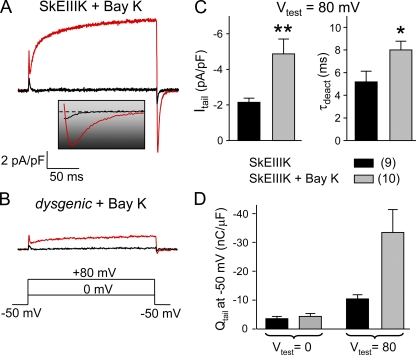
The term excitation-coupled Ca(2+) entry (ECCE) designates the entry of extracellular Ca(2+) into skeletal muscle cells, which occurs in response to prolonged depolarization or pulse trains and depends on the presence of both the 1,4-dihydropyridine receptor (DHPR) in the plasma membrane and the type 1 ryanodine receptor in the sarcoplasmic reticulum (SR) membrane. The ECCE pathway is blocked by pharmacological agents that also block store-operated Ca(2+) entry, is inhibited by dantrolene, is relatively insensitive to the DHP antagonist nifedipine (1 microM), and is permeable to Mn(2+). Here, we have examined the effects of these agents on the L-type Ca(2+) current conducted via the DHPR. We found that the nonspecific cation channel antagonists (2-APB, SKF 96356, La(3+), and Gd(3+)) and dantrolene all inhibited the L-type Ca(2+) current. In addition, complete (>97%) block of the L-type current required concentrations of nifedipine >10 microM. Like ECCE, the L-type Ca(2+) channel displays permeability to Mn(2+) in the absence of external Ca(2+) and produces a Ca(2+) current that persists during prolonged ( approximately 10-second) depolarization. This current appears to contribute to the Ca(2+) transient observed during prolonged KCl depolarization of intact myotubes because (1) the transients in normal myotubes decayed more rapidly in the absence of external Ca(2+); (2) the transients in dysgenic myotubes expressing SkEIIIK (a DHPR alpha(1S) pore mutant thought to conduct only monovalent cations) had a time course like that of normal myotubes in Ca(2+)-free solution and were unaffected by Ca(2+) removal; and (3) after block of SR Ca(2+) release by 200 microM ryanodine, normal myotubes still displayed a large Ca(2+) transient, whereas no transient was detectable in SkEIIIK-expressing dysgenic myotubes. Collectively, these results indicate that the skeletal muscle L-type channel is a major contributor to the Ca(2+) entry attributed to ECCE.
Figures
References
-
- Adams, B.A., T. Tanabe, A. Mikami, S. Numa, and K.G. Beam. 1990. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 346:569–572. - PubMed
-
- Armstrong, C.M., F.M. Bezanilla, and P. Horowicz. 1972. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N′-tetraacetic acid. Biochim. Biophys. Acta. 267:605–608. - PubMed
-
- Beam, K.G., and C. Franzini-Armstrong. 1997. Functional and structural approaches to the study of excitation-contraction coupling. Methods Cell Biol. 52:283–306. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
