

Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline
- PMID: 19116361
- PMCID: PMC2630548
- DOI: 10.2353/ajpath.2009.080565
Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline
Abstract
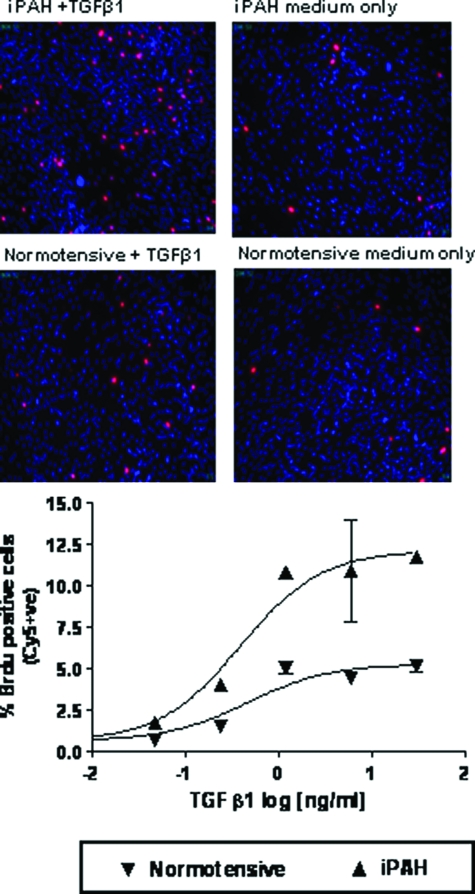
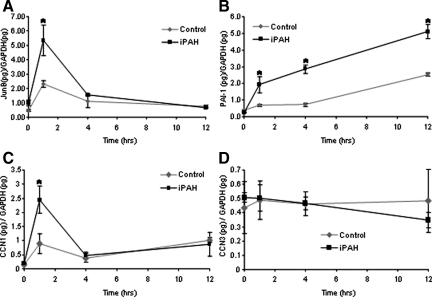
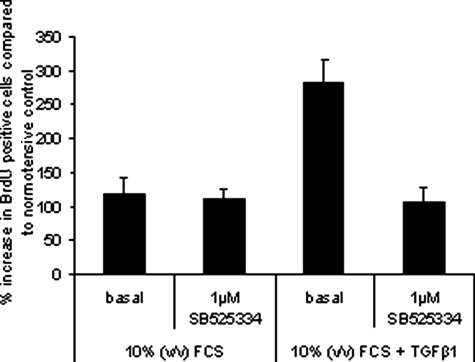
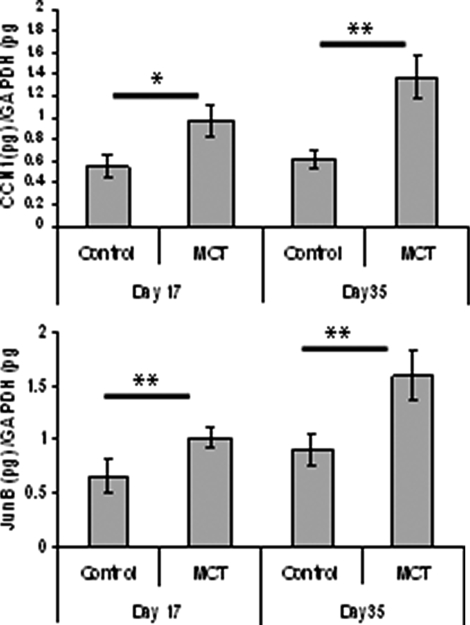
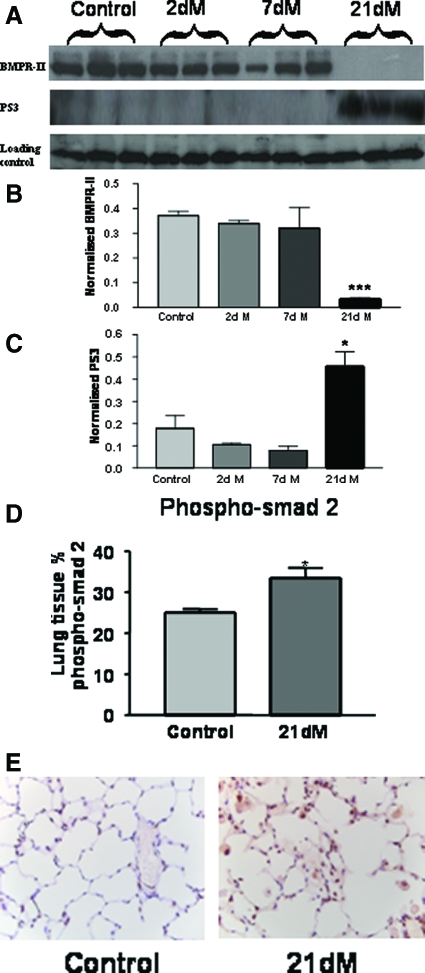
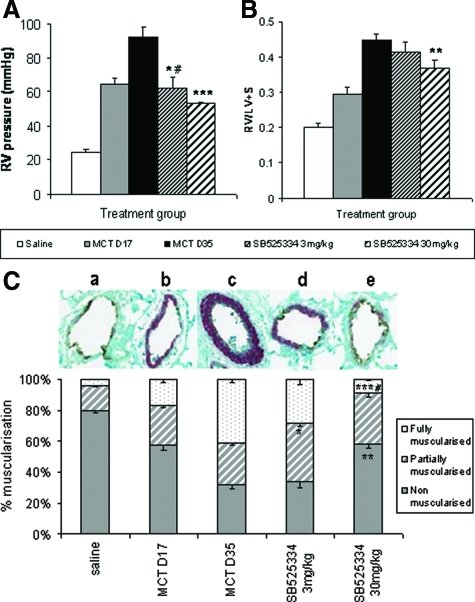
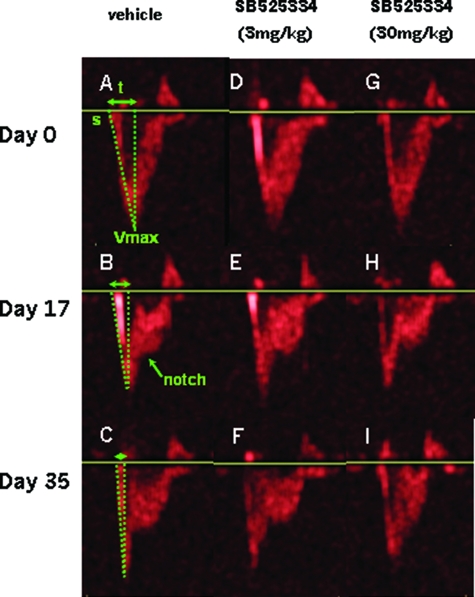
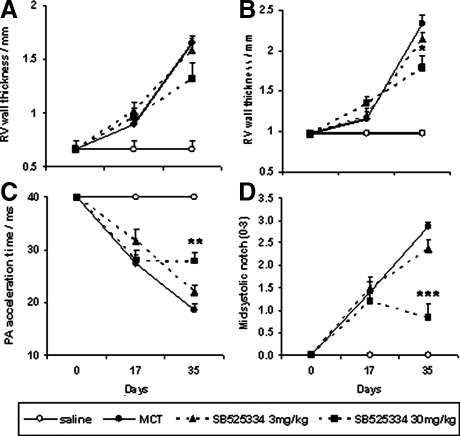
Mutations in the gene for the transforming growth factor (TGF)-beta superfamily receptor, bone morphogenetic protein receptor II, underlie heritable forms of pulmonary arterial hypertension (PAH). Aberrant signaling via TGF-beta receptor I/activin receptor-like kinase 5 may be important for both the development and progression of PAH. We investigated the therapeutic potential of a well-characterized and potent activin receptor-like kinase 5 inhibitor, SB525334 [6-(2-tert-butyl-5-{6-methyl-pyridin-2-yl}-1H-imidazol-4-yl)-quinoxaline] for the treatment of PAH. In this study, we demonstrate that pulmonary artery smooth muscle cells from patients with familial forms of idiopathic PAH exhibit heightened sensitivity to TGF-beta1 in vitro, which can be attenuated after the administration of SB525334. We further demonstrate that SB525334 significantly reverses pulmonary arterial pressure and inhibits right ventricular hypertrophy in a rat model of PAH. Immunohistochemical studies confirmed a significant reduction in pulmonary arteriole muscularization induced by monocrotaline (used experimentally to induce PAH) after treatment of rats with SB525334. Collectively, these data are consistent with a role for the activin receptor-like kinase 5 in the progression of idiopathic PAH and imply that strategies to inhibit activin receptor-like kinase 5 signaling may have therapeutic benefit.
Figures
References
-
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, III, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–84. - PubMed
-
- Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104:790–795. - PubMed
-
- Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth factor-beta signalling in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1340–1348. - PubMed
-
- Jachec W, Foremny A, Domal-Kwiatkowska D, Smolik S, Tomasik A, Mazurek U, Wodniecki J. Expression of TGF-beta1 and its receptor genes (TbetaR I, TbetaR II, and TbetaR III-betaglycan) in peripheral blood leucocytes in patients with idiopathic pulmonary arterial hypertension and Eisenmenger’s syndrome. Int J Mol Med. 2008;21:99–107. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
