

Nephronophthisis: disease mechanisms of a ciliopathy
- PMID: 19118152
- PMCID: PMC2807379
- DOI: 10.1681/ASN.2008050456
Nephronophthisis: disease mechanisms of a ciliopathy
Abstract
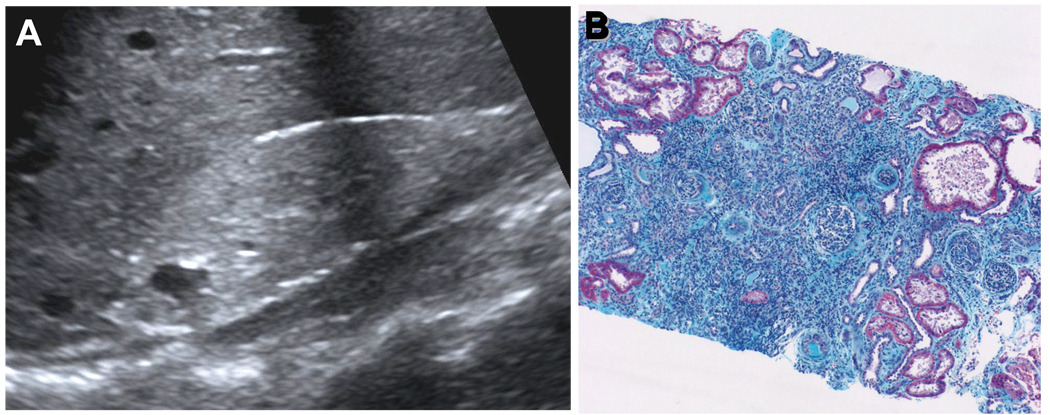
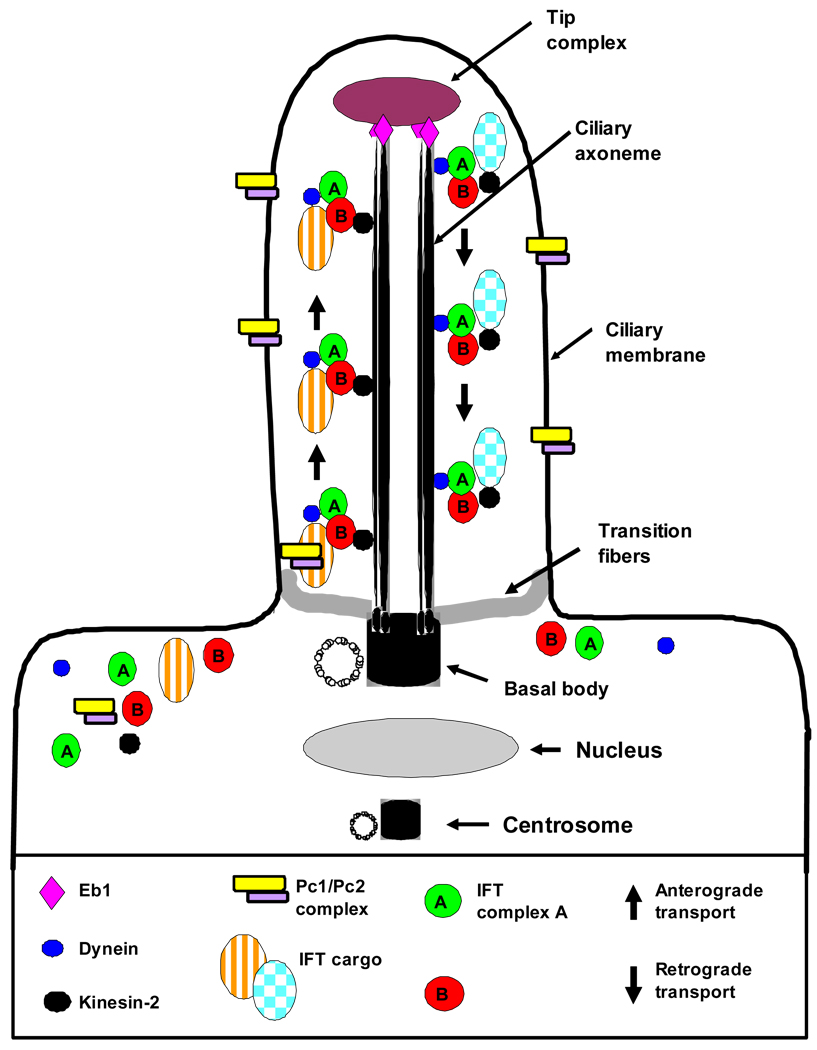
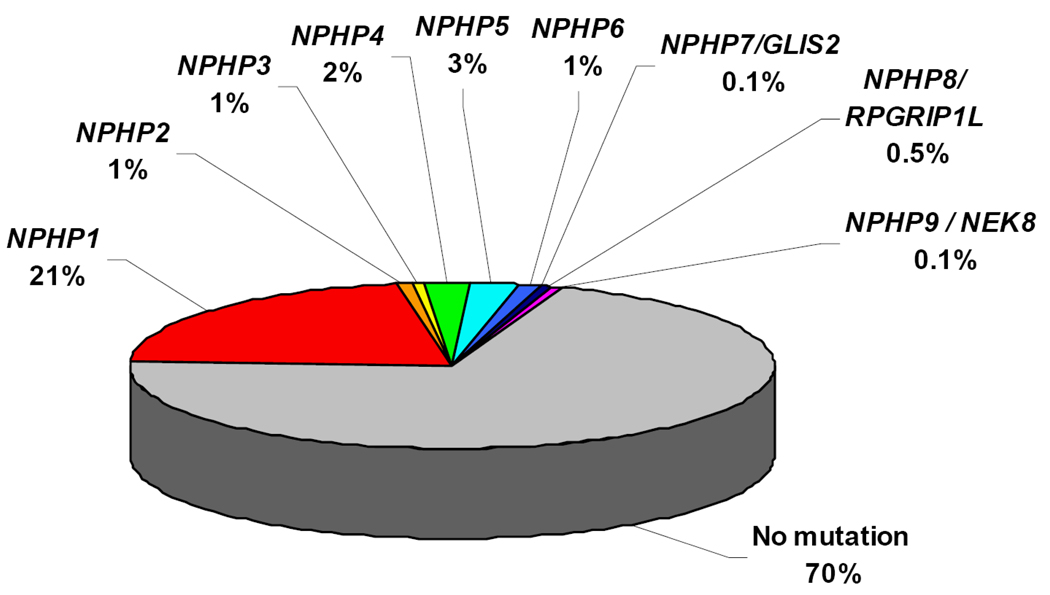
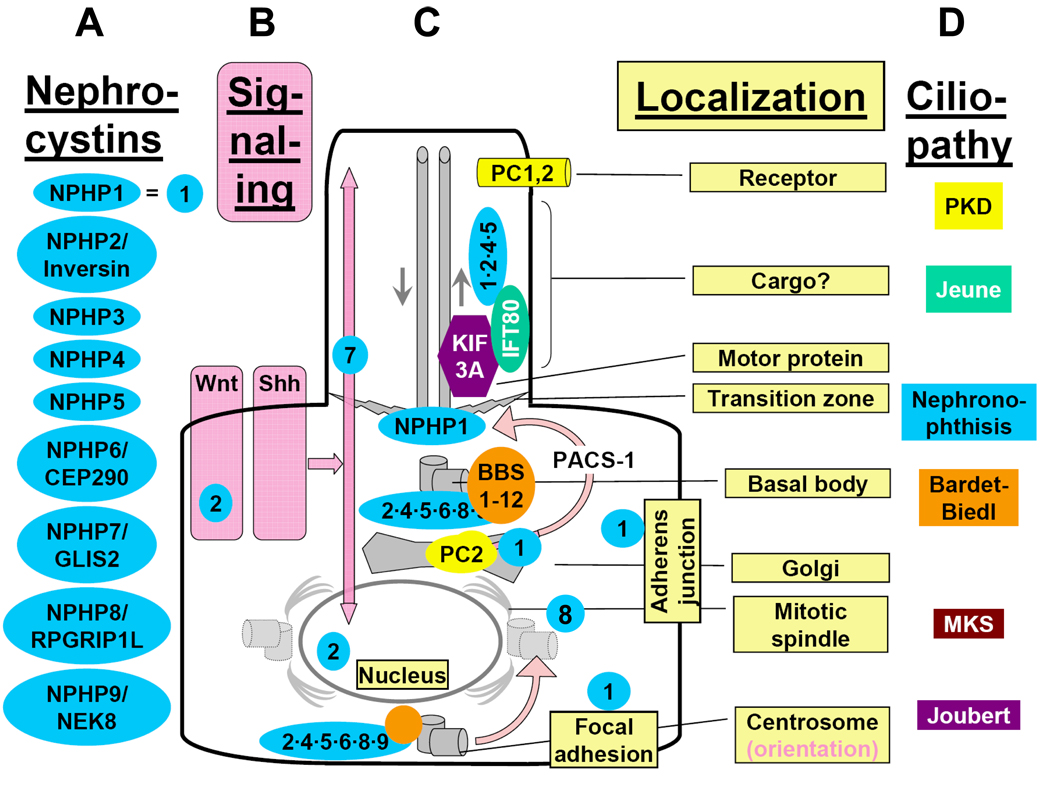
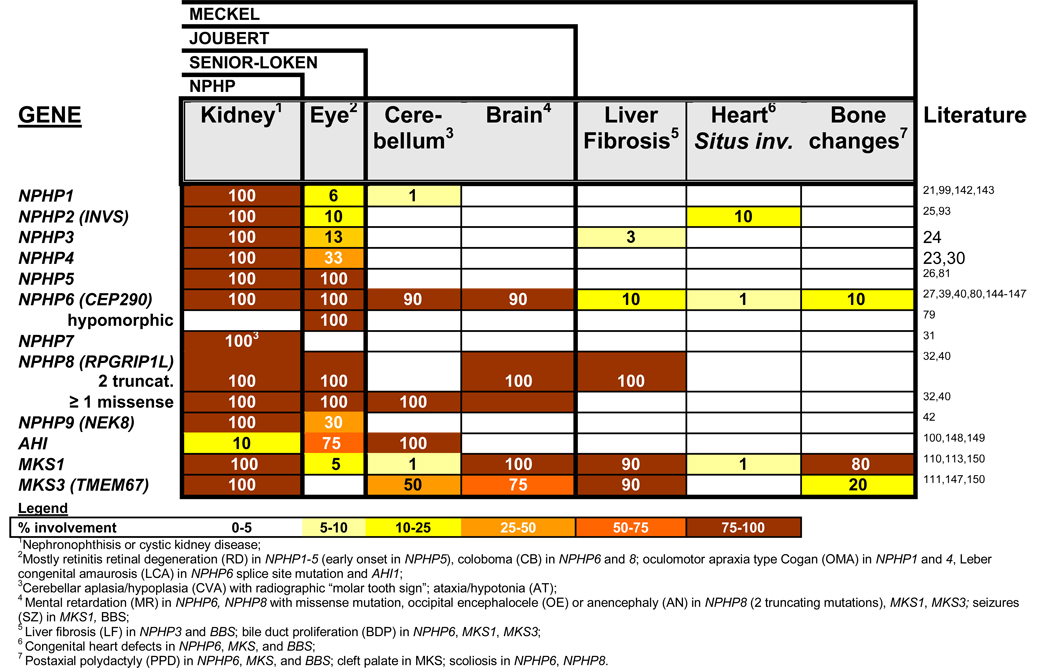
Nephronophthisis (NPHP), a recessive cystic kidney disease, is the most frequent genetic cause of end-stage kidney disease in children and young adults. Positional cloning of nine genes (NPHP1 through 9) and functional characterization of their encoded proteins (nephrocystins) have contributed to a unifying theory that defines cystic kidney diseases as "ciliopathies." The theory is based on the finding that all proteins mutated in cystic kidney diseases of humans or animal models are expressed in primary cilia or centrosomes of renal epithelial cells. Primary cilia are sensory organelles that connect mechanosensory, visual, and other stimuli to mechanisms of epithelial cell polarity and cell-cycle control. Mutations in NPHP genes cause defects in signaling mechanisms that involve the noncanonical Wnt signaling pathway and the sonic hedgehog signaling pathway, resulting in defects of planar cell polarity and tissue maintenance. The ciliary theory explains the multiple organ involvement in NPHP, which includes retinal degeneration, cerebellar hypoplasia, liver fibrosis, situs inversus, and mental retardation. Positional cloning of dozens of unknown genes that cause NPHP will elucidate further signaling mechanisms involved. Nephrocystins are highly conserved in evolution, thereby allowing the use of animal models to develop future therapeutic approaches.
Figures
References
-
- Hildebrandt F, Otto EA. Primary cilia: a unifying pathogenic concept for cystic kidney disease? Nature Reviews Genetics. 2005 - PubMed
-
- Smith C, Graham J. Congenital medullary cysts of the kidneys with severe refractory anemia. Am J Dis Child. 1945;69:369–377.
-
- Fanconi G, Hanhart E, Albertini A. Die familiäre juvenile Nephronophthise. Hel Pediatr Acta. 1951;6:1–49. - PubMed
-
- Hildebrandt F. Juvenile nephronophthisis. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric nephrology. Baltimore: Williams & Wilkins; 1999.
-
- Ala-Mello S, Kivivuori SM, Ronnholm KA, Koskimies O, Siimes MA. Mechanism underlying early anaemia in children with familial juvenile nephronophthisis. Pediatr Nephrol. 1996;10:578–581. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
