

Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K
- PMID: 19122179
- PMCID: PMC2680702
- DOI: 10.1161/CIRCRESAHA.108.182568
Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K
Abstract
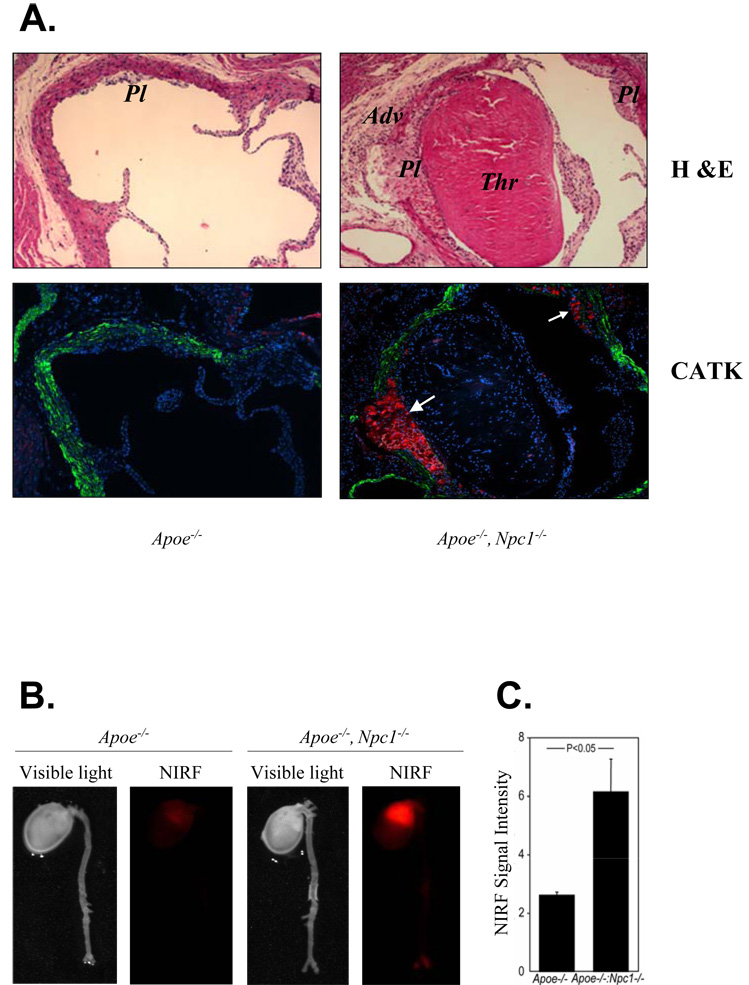
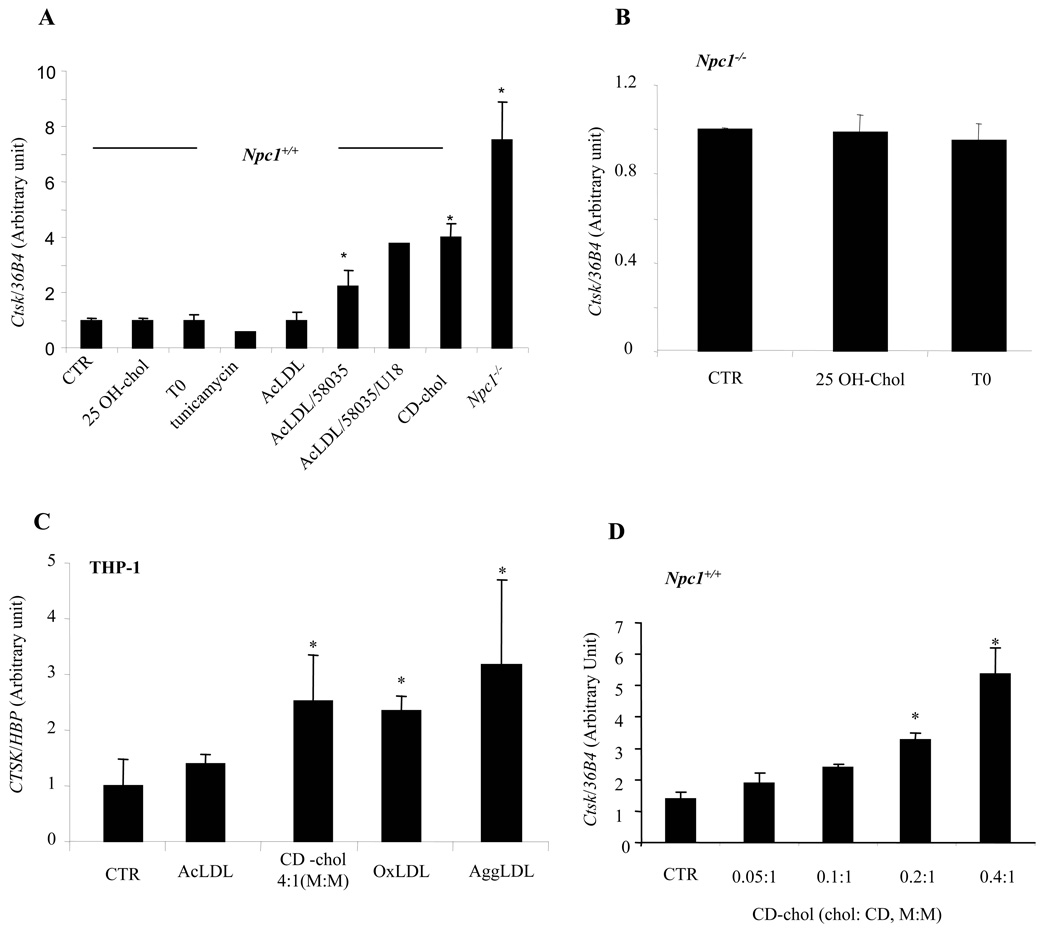
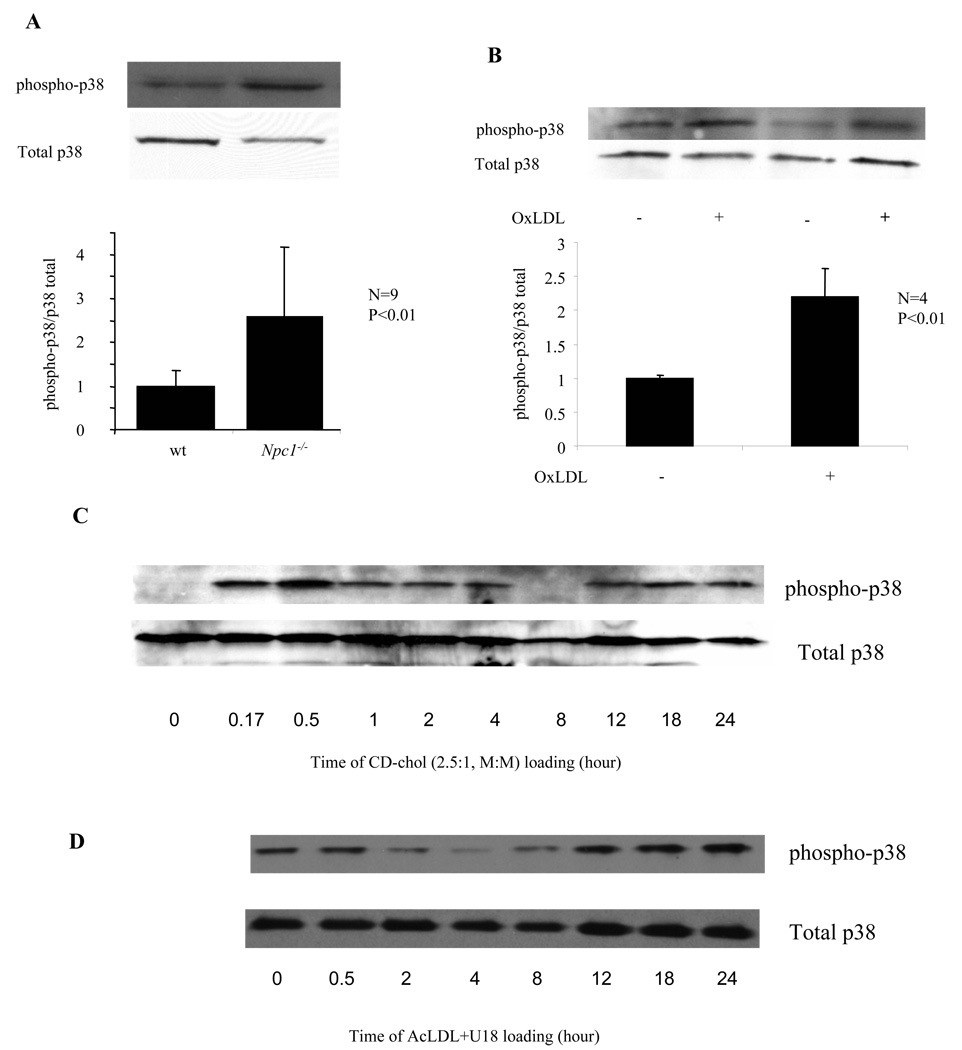
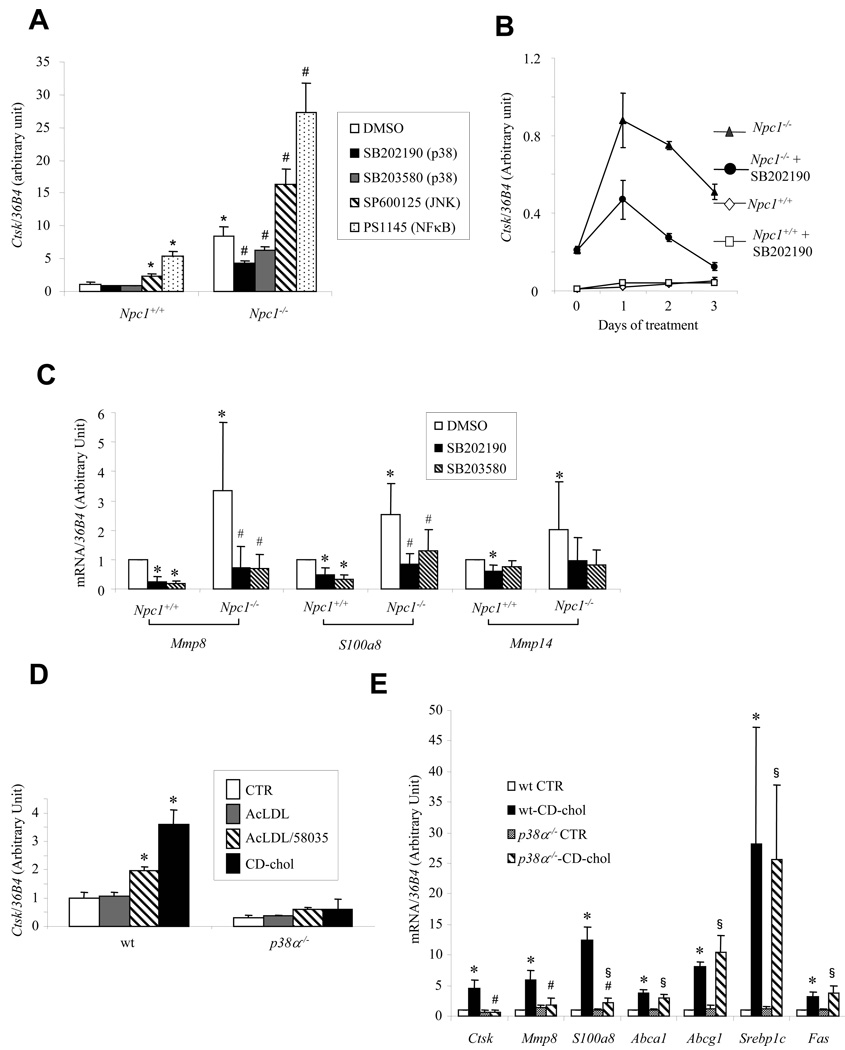
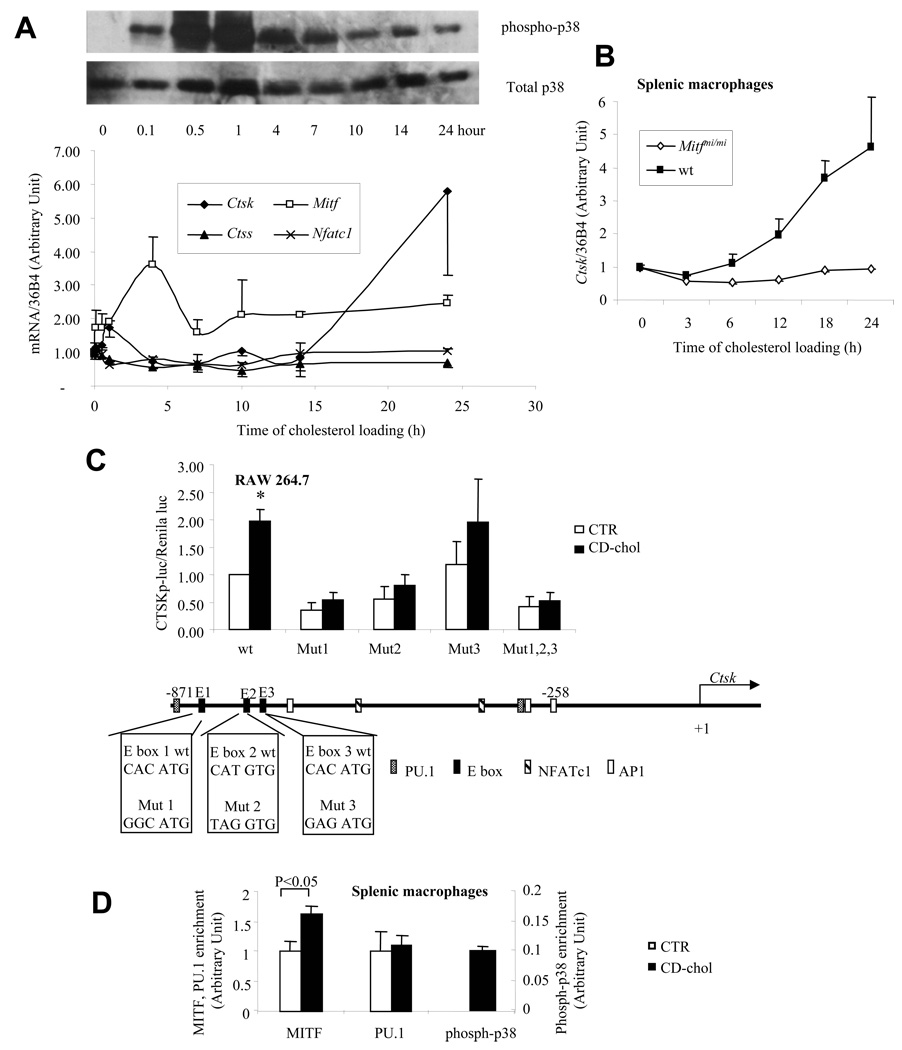
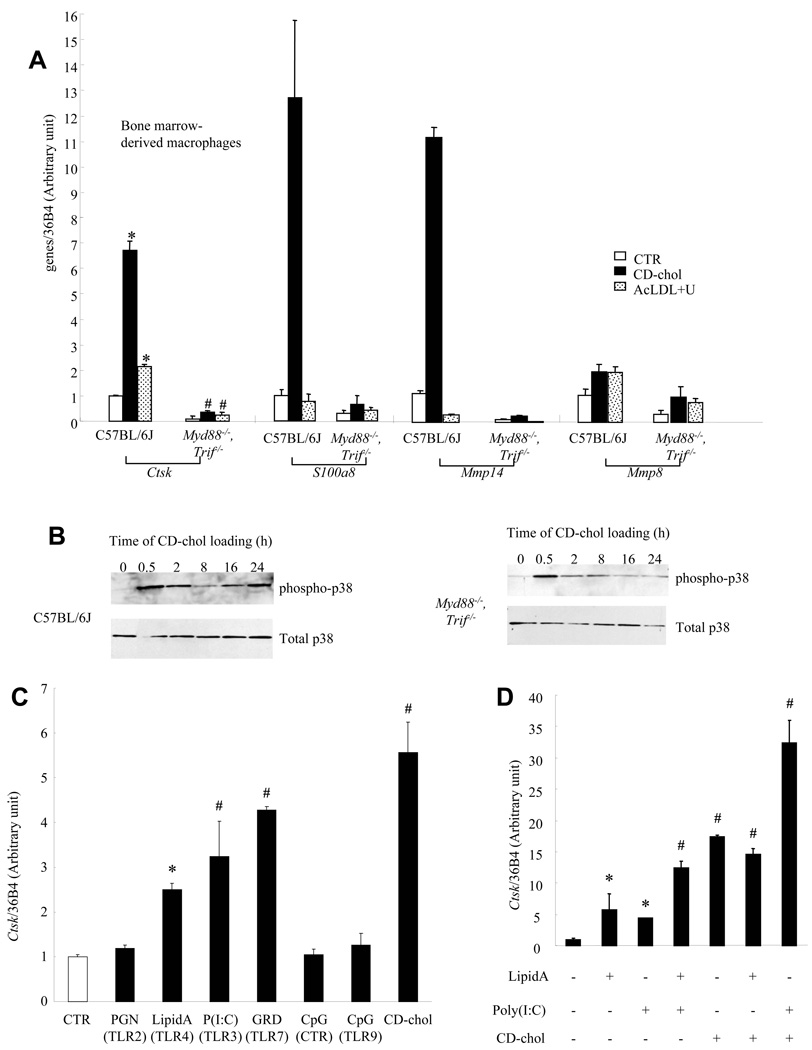
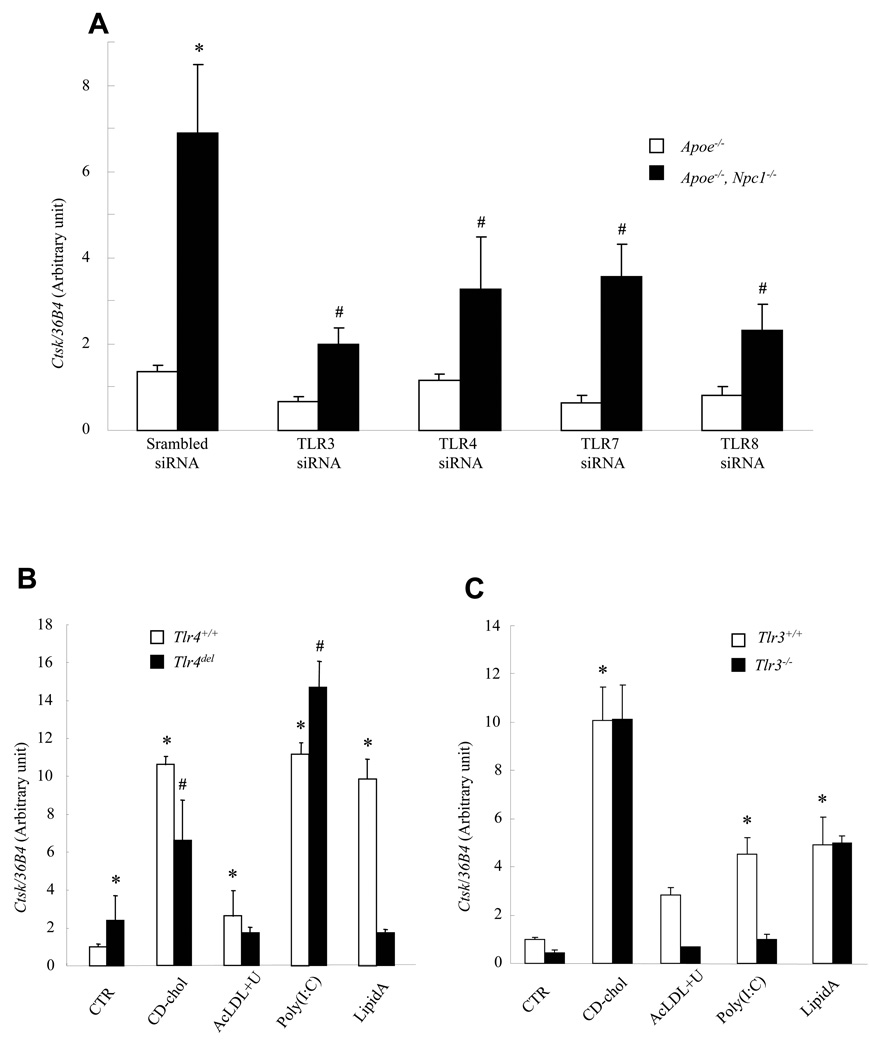
The molecular events linking lipid accumulation in atherosclerotic plaques to complications such as aneurysm formation and plaque disruption are poorly understood. BALB/c-Apoe(-/-) mice bearing a null mutation in the Npc1 gene display prominent medial erosion and atherothrombosis, whereas their macrophages accumulate free cholesterol in late endosomes and show increased cathepsin K (Ctsk) expression. We now show increased cathepsin K immunostaining and increased cysteinyl proteinase activity using near infrared fluorescence imaging over proximal aortas of Apoe(-/-), Npc1(-/-) mice. In mechanistic studies, cholesterol loading of macrophage plasma membranes (cyclodextrin-cholesterol) or endosomal system (AcLDL+U18666A or Npc1 null mutation) activated Toll-like receptor (TLR) signaling, leading to sustained phosphorylation of p38 mitogen-activated protein kinase and induction of p38 targets, including Ctsk, S100a8, Mmp8, and Mmp14. Studies in macrophages from knockout mice showed major roles for TLR4, following plasma membrane cholesterol loading, and for TLR3, after late endosomal loading. TLR signaling via p38 led to phosphorylation and activation of the transcription factor Microphthalmia transcription factor, acting at E-box elements in the Ctsk promoter. These studies suggest that free cholesterol enrichment of either plasma or endosomal membranes in macrophages leads to activation of signaling via various TLRs, prolonged p38 mitogen-activated protein kinase activation, and induction of Mmps, Ctsk, and S100a8, potentially contributing to plaque complications.
Figures
References
-
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. - PubMed
-
- Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation. 2001;103:1718–1720. - PubMed
-
- Rader DJ, Daugherty A. Translating molecular discoveries into new therapies for atherosclerosis. Nature. 2008;451:904–913. - PubMed
-
- Welch CL, Sun Y, Arey BJ, Lemaitre V, Sharma N, Ishibashi M, Sayers S, Li R, Gorelik A, Pleskac N, Collins-Fletcher K, Yasuda Y, Bromme D, D'Armiento JM, Ogletree ML, Tall AR. Spontaneous atherothrombosis and medial degradation in Apoe−/−, Npc1−/− mice. Circulation. 2007;116:2444–2452. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
