

PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls
- PMID: 19122651
- PMCID: PMC2924752
- DOI: 10.1038/nbt.1518
PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls
Abstract
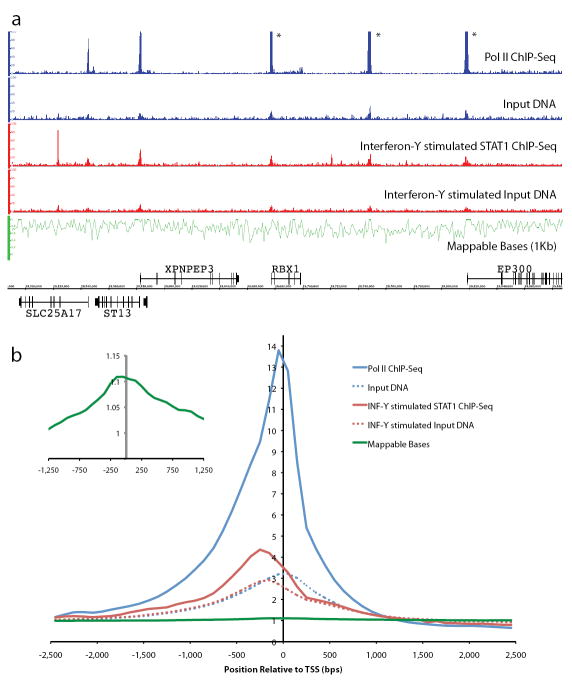
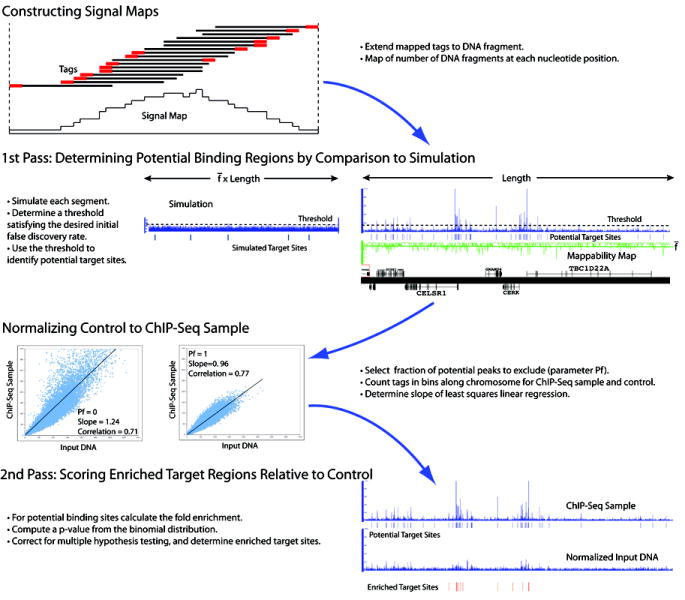
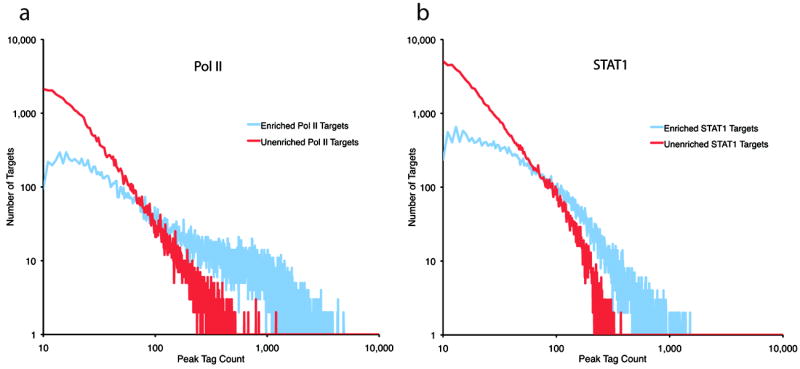
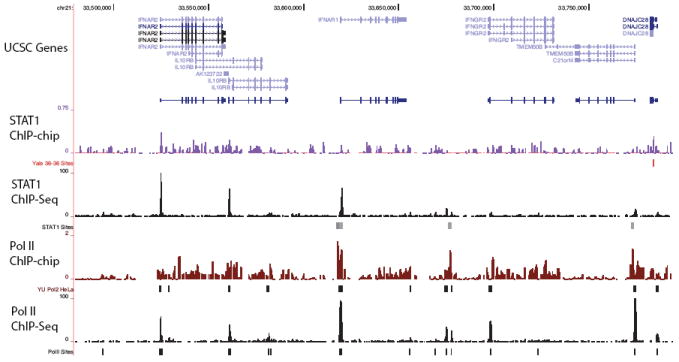
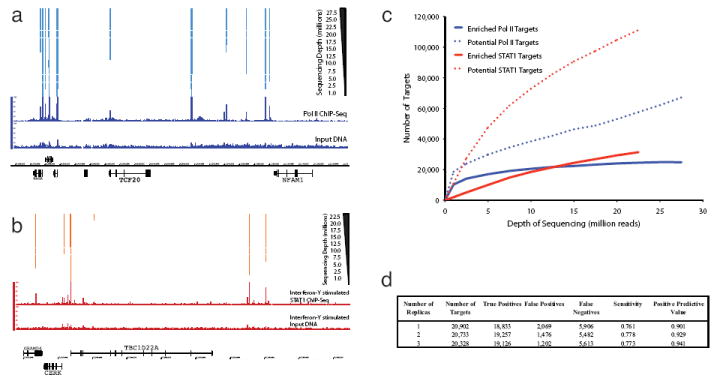
Chromatin immunoprecipitation (ChIP) followed by tag sequencing (ChIP-seq) using high-throughput next-generation instrumentation is fast, replacing chromatin immunoprecipitation followed by genome tiling array analysis (ChIP-chip) as the preferred approach for mapping of sites of transcription-factor binding and chromatin modification. Using two deeply sequenced data sets for human RNA polymerase II and STAT1, each with matching input-DNA controls, we describe a general scoring approach to address unique challenges in ChIP-seq data analysis. Our approach is based on the observation that sites of potential binding are strongly correlated with signal peaks in the control, likely revealing features of open chromatin. We develop a two-pass strategy called PeakSeq to compensate for this. A two-pass strategy compensates for signal caused by open chromatin, as revealed by inclusion of the controls. The first pass identifies putative binding sites and compensates for genomic variation in the 'mappability' of sequences. The second pass filters out sites not significantly enriched compared to the normalized control, computing precise enrichments and significances. Our scoring procedure enables us to optimize experimental design by estimating the depth of sequencing required for a desired level of coverage and demonstrating that more than two replicates provides only a marginal gain in information.
Figures
References
-
- Ren B, et al. Genome-Wide Location and Function of DNA Binding Proteins. Science. 2000;290:2306–2309. - PubMed
-
- Iyer VR, et al. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–8. - PubMed
-
- Horak CE, Snyder M. ChIP-chip: a genomic approach for identifying transcription factor binding sites. Methods Enzymol. 2002;350:469–83. - PubMed
-
- Kim J, et al. Mapping DNA-protein interactions in large genomes by sequence tag analysis of genomic enrichment. Nat Methods. 2005;2:47–53. - PubMed
-
- Wei C, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
