

Proton assisted recoupling and protein structure determination
- PMID: 19123534
- PMCID: PMC2755343
- DOI: 10.1063/1.3036928
Proton assisted recoupling and protein structure determination
Abstract
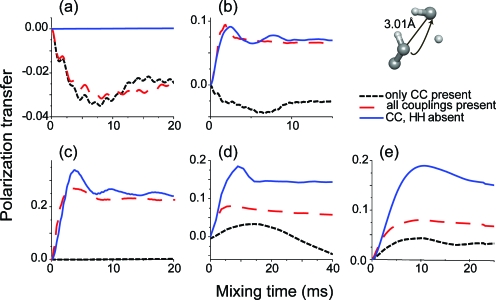
We introduce a homonuclear version of third spin assisted recoupling, a second-order mechanism that can be used for polarization transfer between (13)C or (15)N spins in magic angle spinning (MAS) NMR experiments, particularly at high spinning frequencies employed in contemporary high field MAS experiments. The resulting sequence, which we refer to as proton assisted recoupling (PAR), relies on a cross-term between (1)H-(13)C (or (1)H-(15)N) couplings to mediate zero quantum (13)C-(13)C (or (15)N-(15)N recoupling). In particular, using average Hamiltonian theory we derive an effective Hamiltonian for PAR and show that the transfer is mediated by trilinear terms of the form C(1) (+/-)C(2) (-/+)H(Z) for (13)C-(13)C recoupling experiments (or N(1) (+/-)N(2) (-/+)H(Z) for (15)N-(15)N). We use analytical and numerical simulations to explain the structure of the PAR optimization maps and to delineate the PAR matching conditions. We also detail the PAR polarization transfer dependence with respect to the local molecular geometry and explain the observed reduction in dipolar truncation. Finally, we demonstrate the utility of PAR in structural studies of proteins with (13)C-(13)C spectra of uniformly (13)C, (15)N labeled microcrystalline Crh, a 85 amino acid model protein that forms a domain swapped dimer (MW=2 x 10.4 kDa). The spectra, which were acquired at high MAS frequencies (omega(r)2pi>20 kHz) and magnetic fields (750-900 MHz (1)H frequencies) using moderate rf fields, exhibit numerous cross peaks corresponding to long (up to 6-7 A) (13)C-(13)C distances which are particularly useful in protein structure determination. Using results from PAR spectra we calculate the structure of the Crh protein.
Figures











References
-
- Munowitz M. G., Griffin R. G., Bodenhausen G., and Huang T. H., J. Am. Chem. Soc. JACSAT10.1021/ja00400a007 103, 2529 (1981) - DOI
- Munowitz M. G. and Griffin R. G., J. Chem. Phys. JCPSA610.1063/1.443386 76, 2848 (1982) - DOI
- Raleigh D. P., Levitt M. H., and Griffin R. G., Chem. Phys. Lett. CHPLBC10.1016/0009-2614(88)85051-6 146, 71 (1988) - DOI
- Raleigh D. P., Creuzet F., Gupta S. K. D., Levitt M. H., and Griffin R. G., J. Am. Chem. Soc. JACSAT10.1021/ja00194a057 111, 4502 (1989) - DOI
- Tycko R. and Dabbagh G., Chem. Phys. Lett. CHPLBC10.1016/0009-2614(90)87235-J 173, 461 (1990) - DOI
- Tycko R. and Dabbagh G., J. Am. Chem. Soc. JACSAT10.1021/ja00025a003 113, 9444 (1991) - DOI
- Bayro M. J., Ramachandran R., Caporini M. A., Eddy M. T., and Griffin R. G., J. Chem. Phys. 128, (2008) - PubMed
- Brinkmann A., Eden M., and Levitt M. H., J. Chem. Phys. JCPSA610.1063/1.481458 112, 8539 (2000) - DOI
- Brinkmann A., Eden M., and Levitt M. H., J. Chem. Phys. JCPSA610.1063/1.1377031 115, 357 (2001). - DOI
-
- Gullion T. and Schaefer J., J. Magn. Reson. 81, 196 (1989).
-
- Bennett A. E., Ok J. H., Griffin R. G., and Vega S., J. Chem. Phys. JCPSA610.1063/1.462267 96, 8624 (1992). - DOI
-
- Bennett A. E., Rienstra C. M., Griffiths J. M., Zhen W. G., Lansbury P. T., and Griffin R. G., J. Chem. Phys. JCPSA610.1063/1.476420 108, 9463 (1998). - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous