

Reassessing the amyloid cascade hypothesis of Alzheimer's disease
- PMID: 19124085
- PMCID: PMC2680505
- DOI: 10.1016/j.biocel.2008.12.015
Reassessing the amyloid cascade hypothesis of Alzheimer's disease
Abstract
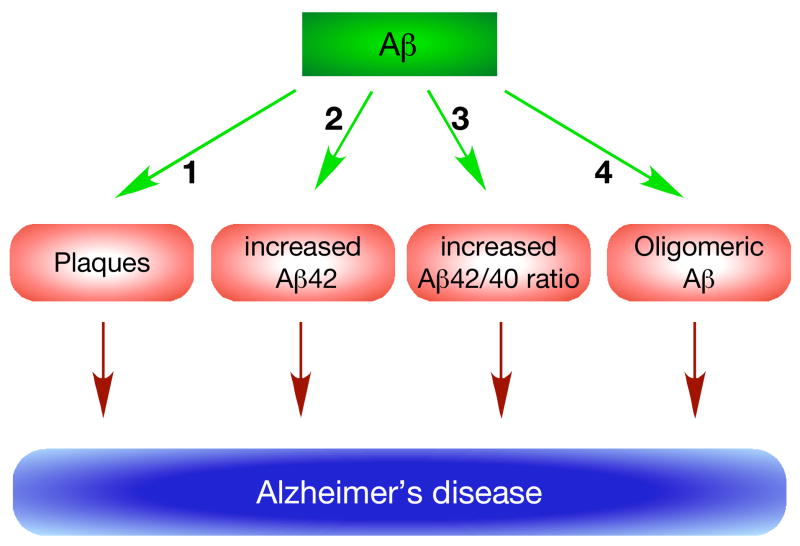
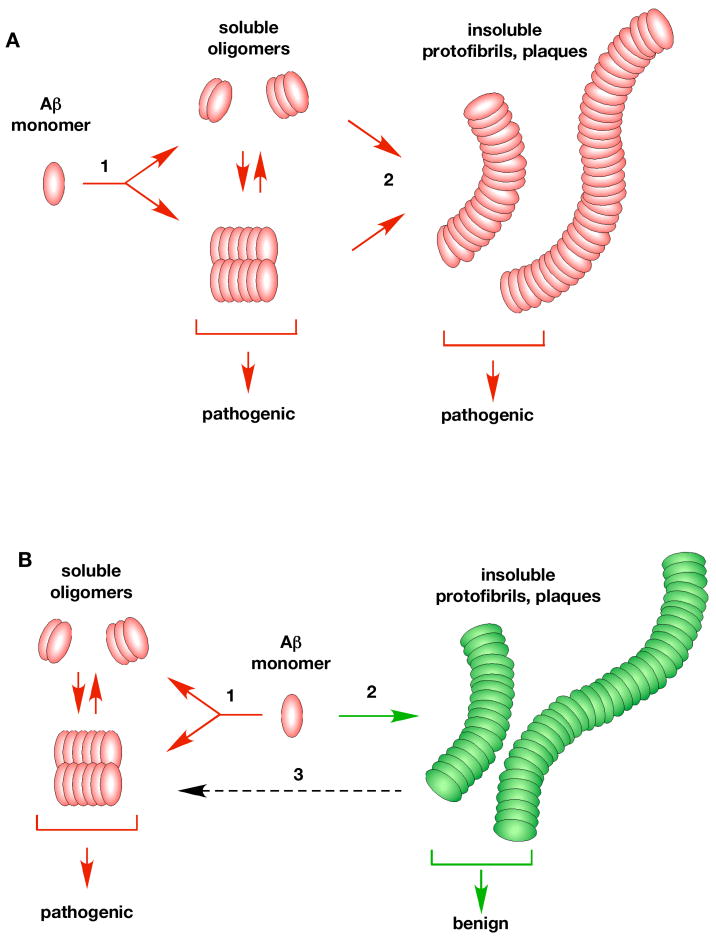
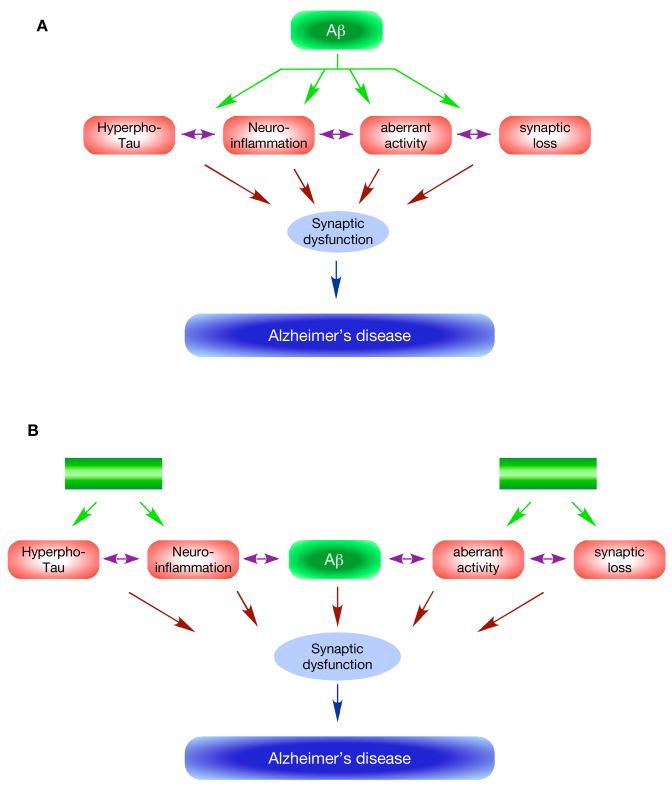
Since its inception, the amyloid cascade hypothesis has dominated the field of Alzheimer's disease (AD) research and has provided the intellectual framework for therapeutic intervention. Although the details of the hypothesis continue to evolve, its core principle has remained essentially unaltered. It posits that the amyloid-beta peptides, derived from amyloid precursor protein (APP), are the root cause of AD. Substantial genetic and biochemical data support this view, and yet a number of findings also run contrary to its tenets. The presence of familial AD mutations in APP and presenilins, demonstration of Abeta toxicity, and studies in mouse models of AD all support the hypothesis, whereas the presence of Abeta plaques in normal individuals, the uncertain nature of the pathogenic Abeta species, and repeated disappointments with Abeta-centered therapeutic trials are inconsistent with the hypothesis. The current state of knowledge does not prove nor disprove the amyloid hypothesis, but rather points to the need for its reassessment. A view that Abeta is one of the factors, as opposed to the factor, that causes AD is more consistent with the present knowledge, and is more likely to promote comprehensive and effective therapeutic strategies.
Figures
References
-
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. - PubMed
-
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De Strooper B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem. 2006;96:732–742. - PubMed
-
- Bitan G, Fradinger EA, Spring SM, Teplow DB. Neurotoxic protein oligomers--what you see is not always what you get. Amyloid. 2005;12:88–95. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
