

Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus
- PMID: 19124763
- PMCID: PMC2745922
- DOI: 10.4049/jimmunol.182.2.1192
Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus
Abstract
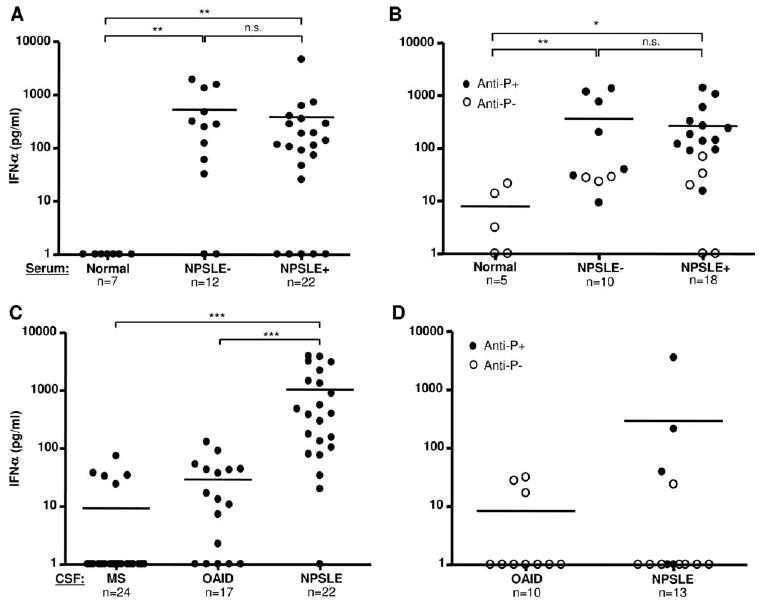
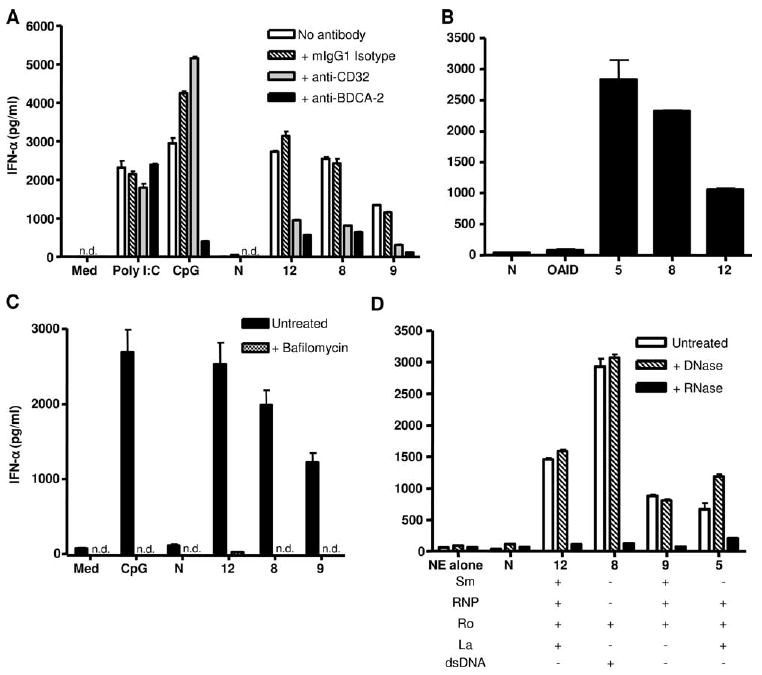
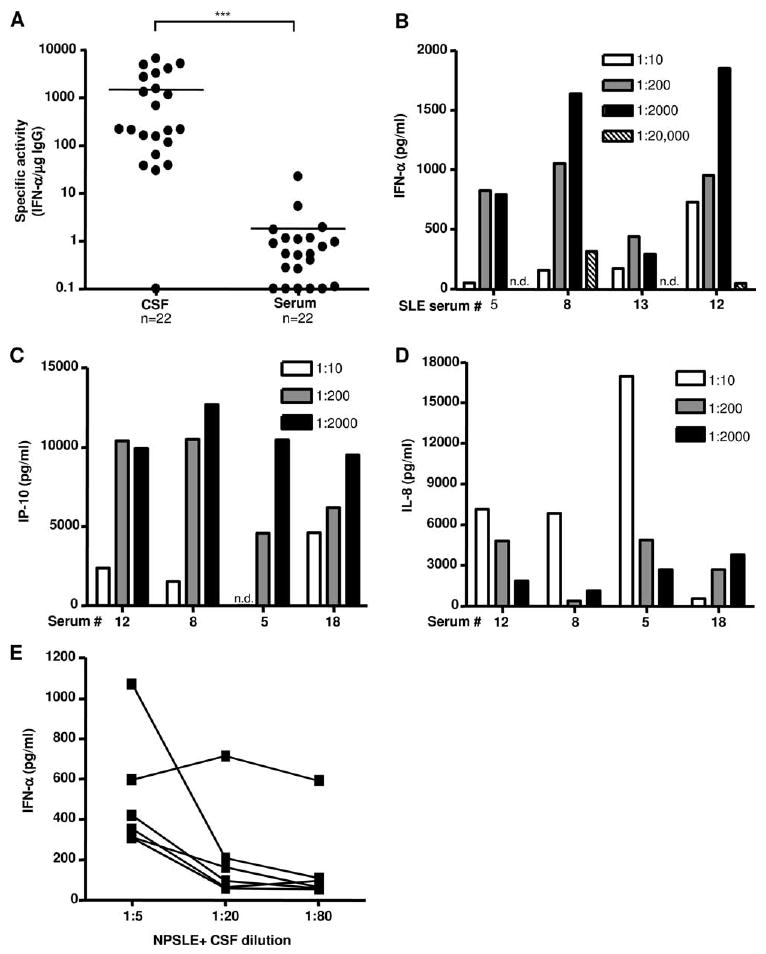
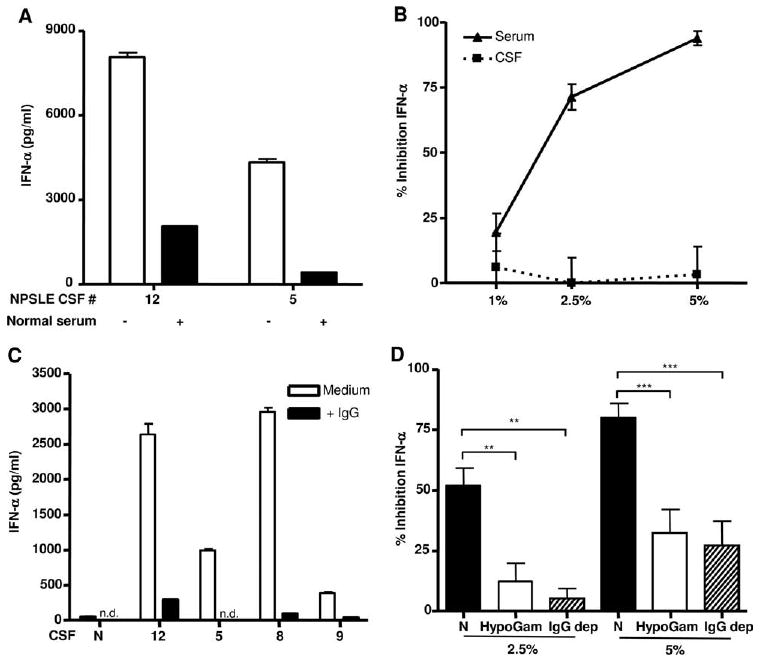
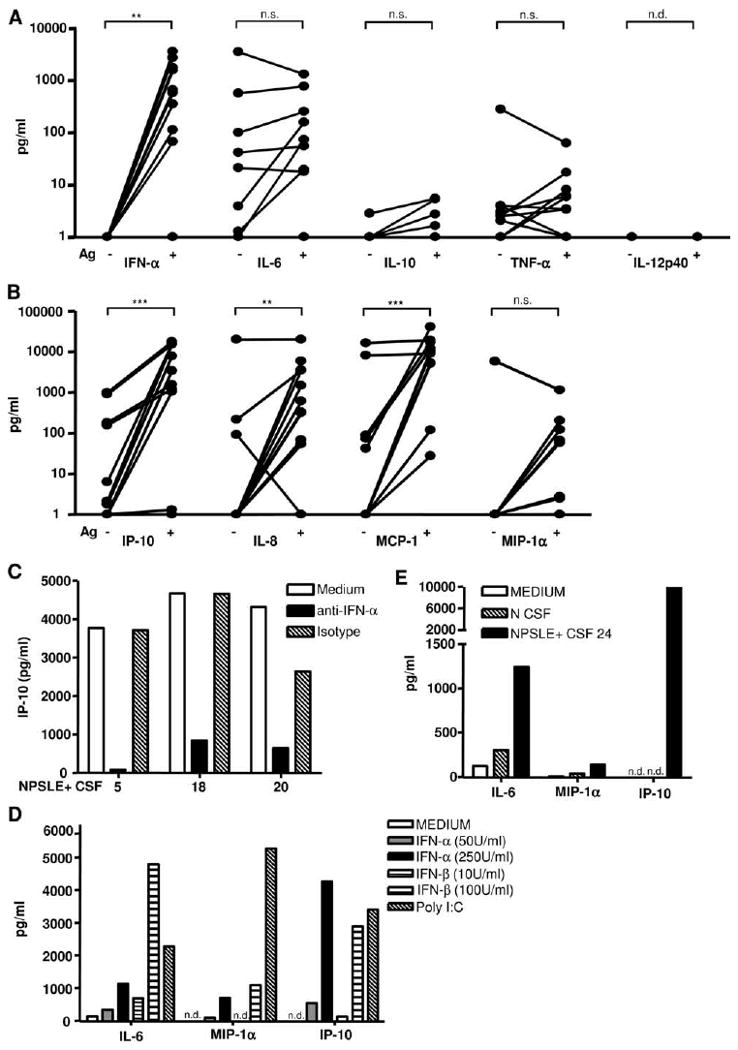
Neuropsychiatric disease in systemic lupus erythematosus (NPSLE) is a poorly understood, but potentially fatal, disease manifestation. A pathogenetic role for autoantibodies is suspected, but the mechanism is unclear. Since immune complexes in SLE can stimulate IFN-alpha and there is strong evidence in humans and in mice that IFN-alpha can cause neuropsychiatric manifestations, we asked whether NPSLE patient serum and/or cerebrospinal fluid (CSF) contain abnormally high IFN-alpha-inducing activity. In a bioassay containing plasmacytoid dendritic cells and a source of Ag, NPSLE CSF induced significantly higher IFN-alpha compared with CSF from patients with multiple sclerosis or other autoimmune disease controls. When normalized for IgG concentration, NPSLE CSF was 800-fold more potent at inducing IFN-alpha compared with paired serum due to inhibitors present in serum. Analysis of Ig-deficient patient serum, depletion of IgG from normal serum, as well as addition of purified IgG to NPSLE CSF and serum in the bioassays revealed that one inhibitor was contained within the IgG fraction itself. In addition to IFN-alpha, immune complexes formed by CSF autoantibodies produced significantly increased levels of IFN-gamma-inducible protein 10 (IP-10/CXCL), IL-8, and MCP-1, all of which have been reported to be elevated in CSF from NPSLE patients. Taken together, these findings are consistent with a two-step model of NPSLE whereby CSF autoantibodies bind to Ags released by neurocytotoxic Abs or other brain cell injury, and the resulting immune complexes stimulate IFN-alpha and proinflammatory cytokines and chemokines.
Conflict of interest statement
Figures
References
-
- Johnson RT, Richardson EP. The neurological manifestations of systemic lupus erythematosus. Medicine. 1968;47:337–369. - PubMed
-
- Hanly JG. Neuropsychiatric lupus. Rheum Dis Clin North Am. 2005;31:273–298. - PubMed
-
- Zandman-Goddard G, Chapman J, Shoenfeld Y. Autoantibodies involved in neuropsychiatric SLE and antiphospholipid syndrome. Semin Arthritis Rheum. 2007;36:297–315. - PubMed
-
- Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HA, 3rd, Koffler D. Intrathecal IgG synthesis and blood-brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med. 1983;74:837–844. - PubMed
-
- Small P, Mass MF, Kohler PF, Harbeck RJ. Central nervous system involvement in SLE: diagnostic profile and clinical features. Arthritis Rheum. 1977;20:869–878. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
