

Cardiovascular actions of neurotrophins
- PMID: 19126759
- PMCID: PMC2836529
- DOI: 10.1152/physrev.00007.2008
Cardiovascular actions of neurotrophins
Abstract
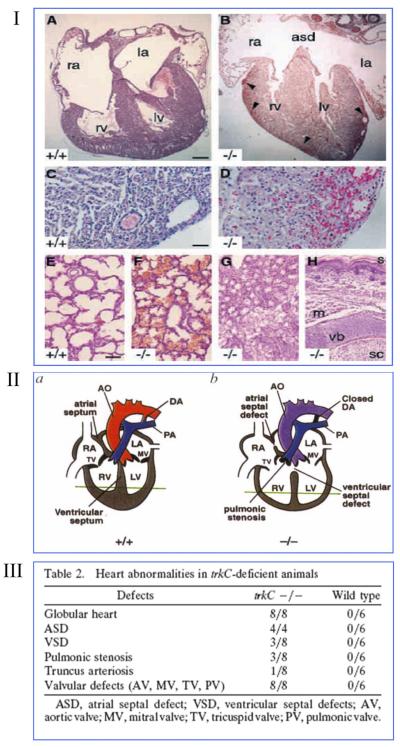
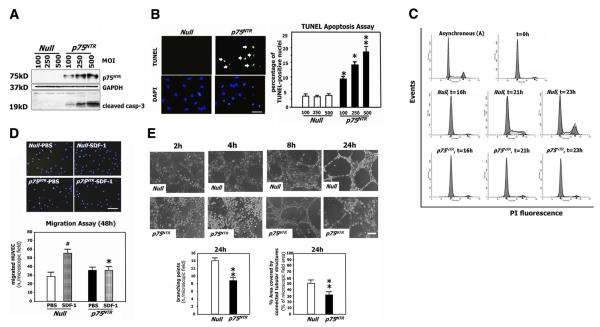
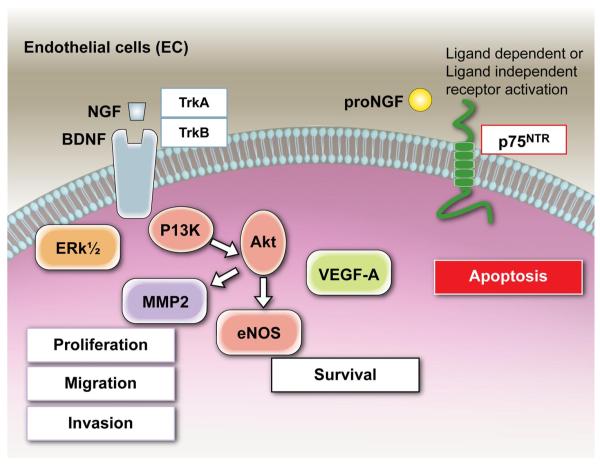
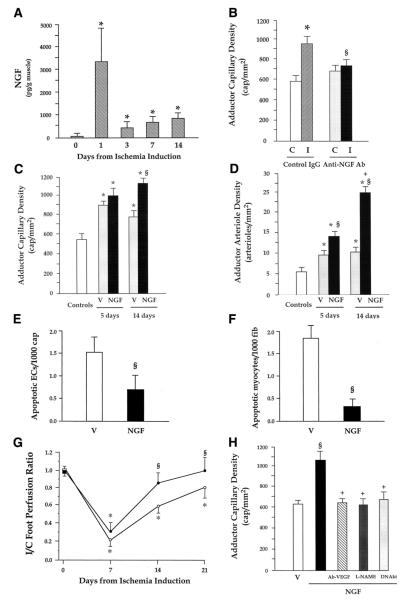
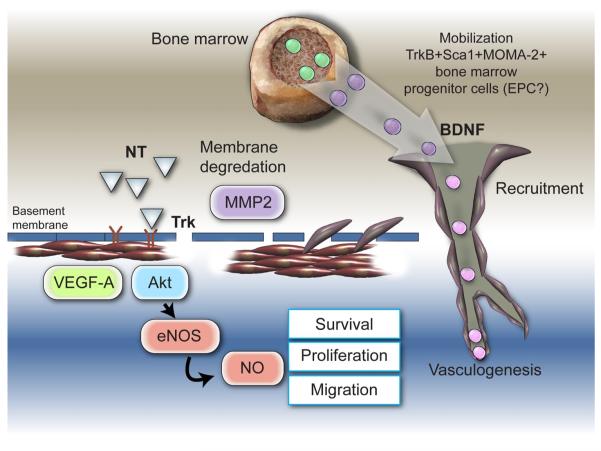
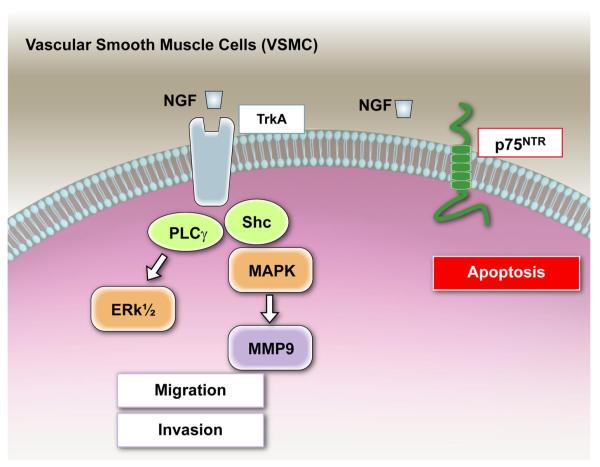
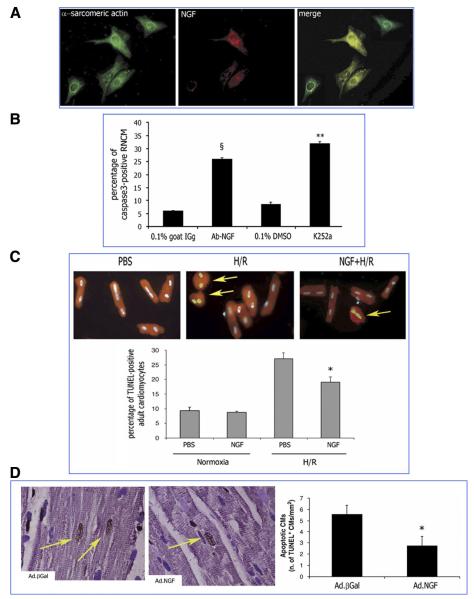
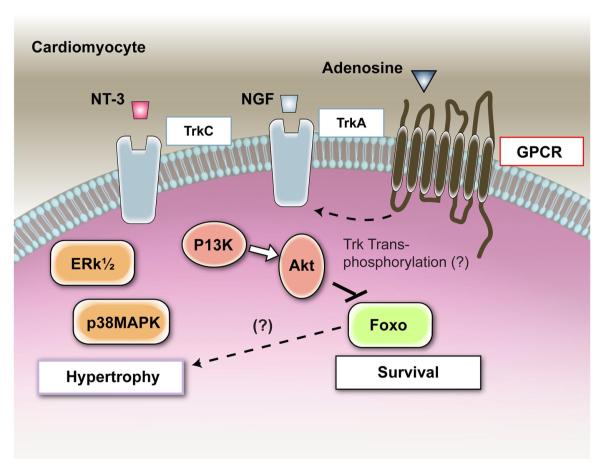
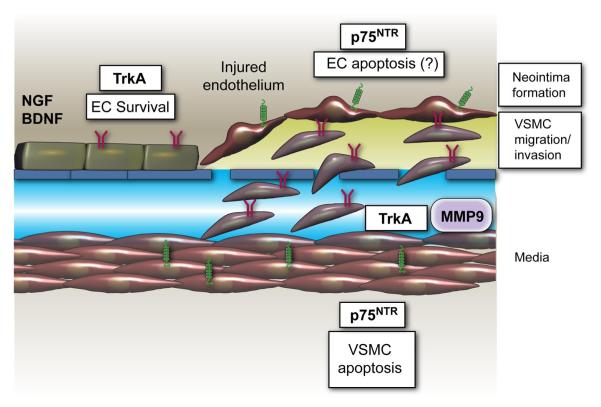
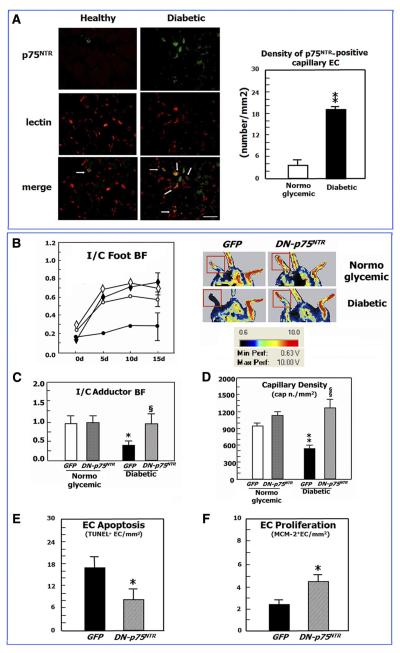
Neurotrophins were christened in consideration of their actions on the nervous system and, for a long time, they were the exclusive interest of neuroscientists. However, more recently, this family of proteins has been shown to possess essential cardiovascular functions. During cardiovascular development, neurotrophins and their receptors are essential factors in the formation of the heart and critical regulator of vascular development. Postnatally, neurotrophins control the survival of endothelial cells, vascular smooth muscle cells, and cardiomyocytes and regulate angiogenesis and vasculogenesis, by autocrine and paracrine mechanisms. Recent studies suggest the capacity of neurotrophins, via their tropomyosin-kinase receptors, to promote therapeutic neovascularization in animal models of hindlimb ischemia. Conversely, the neurotrophin low-affinity p75(NTR) receptor induces apoptosis of endothelial cells and vascular smooth muscle cells and impairs angiogenesis. Finally, nerve growth factor looks particularly promising in treating microvascular complications of diabetes or reducing cardiomyocyte apoptosis in the infarcted heart. These seminal discoveries have fuelled basic and translational research and thus opened a new field of investigation in cardiovascular medicine and therapeutics. Here, we review recent progress on the molecular signaling and roles played by neurotrophins in cardiovascular development, function, and pathology, and we discuss therapeutic potential of strategies based on neurotrophin manipulation.
Figures
References
-
- Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. - PubMed
-
- Anand P. Neurotrophic factors and their receptors in human sensory neuropathies. Prog Brain Res. 2004;146:477–492. - PubMed
-
- Anand P, Foley P, Navsaria HA, Sinicropi D, Williams-Chestnut RE, Leigh IM. Nerve growth factor levels in cultured human skin cells: effect of gestation and viral transformation. Neurosci Lett. 1995;184:157–160. - PubMed
-
- Anand P, Terenghi G, Warner G, Kopelman P, Williams-Chestnut RE, Sinicropi DV. The role of endogenous nerve growth factor in human diabetic neuropathy. Nat Med. 1996;2:703–707. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials