

Control of stochastic gene expression by host factors at the HIV promoter
- PMID: 19132086
- PMCID: PMC2607019
- DOI: 10.1371/journal.ppat.1000260
Control of stochastic gene expression by host factors at the HIV promoter
Abstract
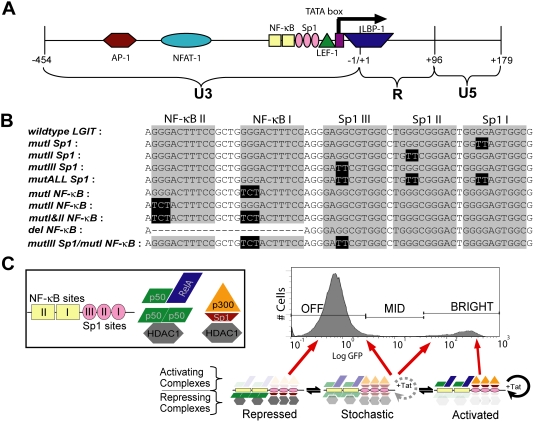
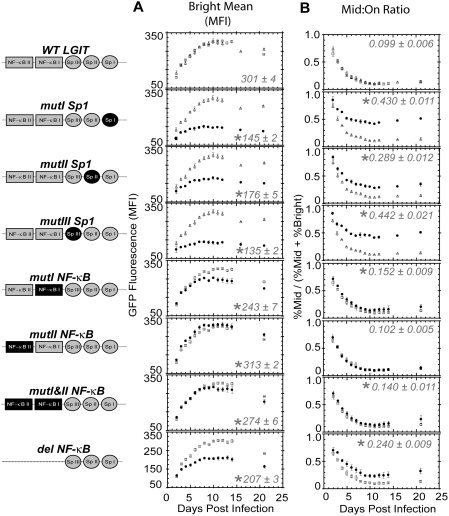
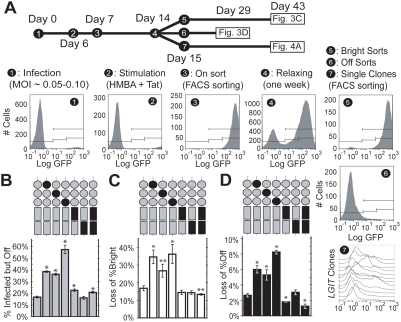
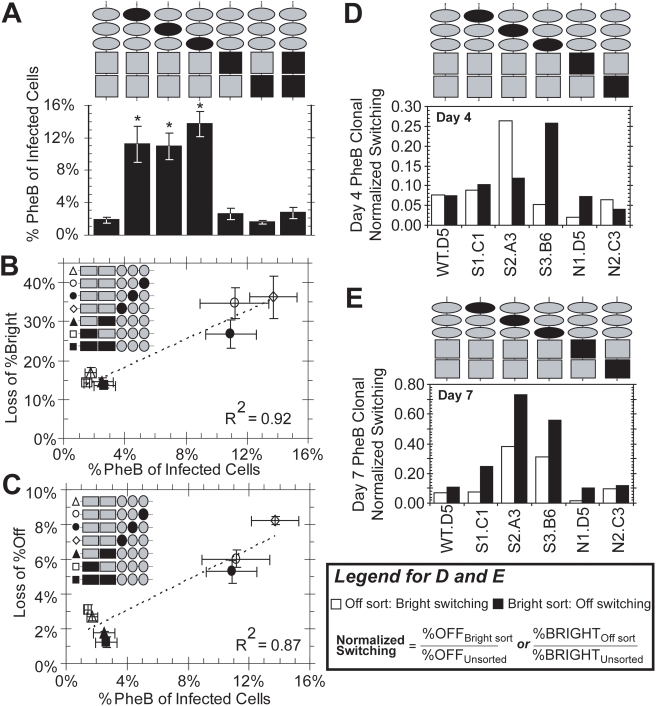
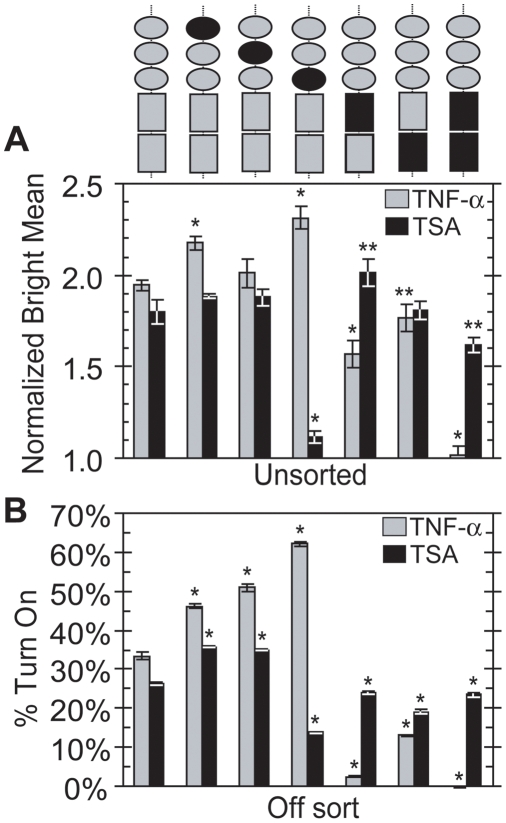
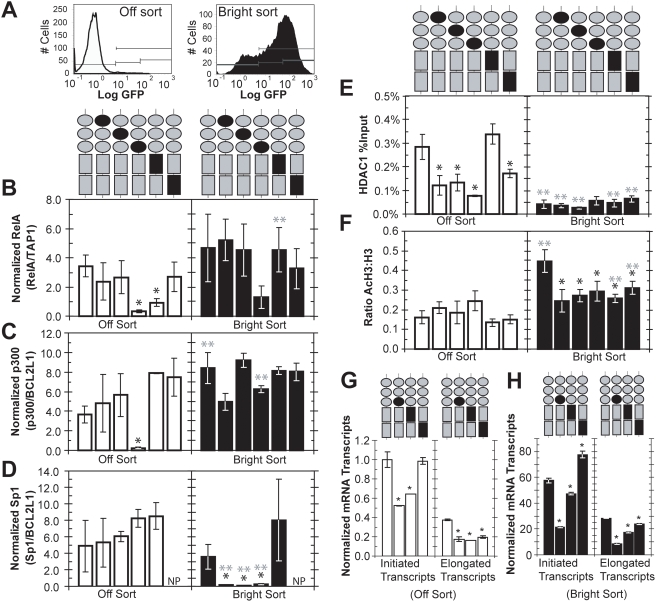
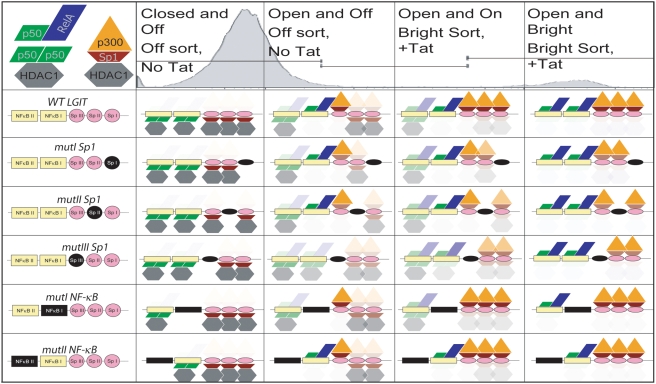
The HIV promoter within the viral long terminal repeat (LTR) orchestrates many aspects of the viral life cycle, from the dynamics of viral gene expression and replication to the establishment of a latent state. In particular, after viral integration into the host genome, stochastic fluctuations in viral gene expression amplified by the Tat positive feedback loop can contribute to the formation of either a productive, transactivated state or an inactive state. In a significant fraction of cells harboring an integrated copy of the HIV-1 model provirus (LTR-GFP-IRES-Tat), this bimodal gene expression profile is dynamic, as cells spontaneously and continuously flip between active (Bright) and inactive (Off) expression modes. Furthermore, these switching dynamics may contribute to the establishment and maintenance of proviral latency, because after viral integration long delays in gene expression can occur before viral transactivation. The HIV-1 promoter contains cis-acting Sp1 and NF-kappaB elements that regulate gene expression via the recruitment of both activating and repressing complexes. We hypothesized that interplay in the recruitment of such positive and negative factors could modulate the stability of the Bright and Off modes and thereby alter the sensitivity of viral gene expression to stochastic fluctuations in the Tat feedback loop. Using model lentivirus variants with mutations introduced in the Sp1 and NF-kappaB elements, we employed flow cytometry, mRNA quantification, pharmacological perturbations, and chromatin immunoprecipitation to reveal significant functional differences in contributions of each site to viral gene regulation. Specifically, the Sp1 sites apparently stabilize both the Bright and the Off states, such that their mutation promotes noisy gene expression and reduction in the regulation of histone acetylation and deacetylation. Furthermore, the NF-kappaB sites exhibit distinct properties, with kappaB site I serving a stronger activating role than kappaB site II. Moreover, Sp1 site III plays a particularly important role in the recruitment of both p300 and RelA to the promoter. Finally, analysis of 362 clonal cell populations infected with the viral variants revealed that mutations in any of the Sp1 sites yield a 6-fold higher frequency of clonal bifurcation compared to that of the wild-type promoter. Thus, each Sp1 and NF-kappaB site differentially contributes to the regulation of viral gene expression, and Sp1 sites functionally "dampen" transcriptional noise and thereby modulate the frequency and maintenance of this model of viral latency. These results may have biomedical implications for the treatment of HIV latency.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–593. - PubMed
-
- Gatignol A, Buckler-White A, Berkhout B, Jeang KT. Characterization of a human TAR RNA-binding protein that activates the HIV-1 LTR. Science. 1991;251:1597–1600. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
