

Progressive familial intrahepatic cholestasis
- PMID: 19133130
- PMCID: PMC2647530
- DOI: 10.1186/1750-1172-4-1
Progressive familial intrahepatic cholestasis
Abstract
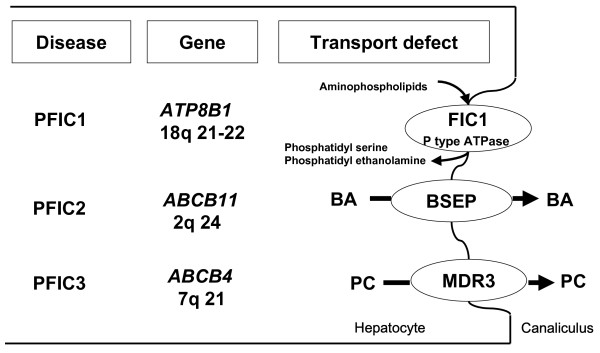
Progressive familial intrahepatic cholestasis (PFIC) refers to heterogeneous group of autosomal recessive disorders of childhood that disrupt bile formation and present with cholestasis of hepatocellular origin. The exact prevalence remains unknown, but the estimated incidence varies between 1/50,000 and 1/100,000 births. Three types of PFIC have been identified and related to mutations in hepatocellular transport system genes involved in bile formation. PFIC1 and PFIC2 usually appear in the first months of life, whereas onset of PFIC3 may also occur later in infancy, in childhood or even during young adulthood. Main clinical manifestations include cholestasis, pruritus and jaundice. PFIC patients usually develop fibrosis and end-stage liver disease before adulthood. Serum gamma-glutamyltransferase (GGT) activity is normal in PFIC1 and PFIC2 patients, but is elevated in PFIC3 patients. Both PFIC1 and PFIC2 are caused by impaired bile salt secretion due respectively to defects in ATP8B1 encoding the FIC1 protein, and in ABCB11 encoding the bile salt export pump protein (BSEP). Defects in ABCB4, encoding the multi-drug resistant 3 protein (MDR3), impair biliary phospholipid secretion resulting in PFIC3. Diagnosis is based on clinical manifestations, liver ultrasonography, cholangiography and liver histology, as well as on specific tests for excluding other causes of childhood cholestasis. MDR3 and BSEP liver immunostaining, and analysis of biliary lipid composition should help to select PFIC candidates in whom genotyping could be proposed to confirm the diagnosis. Antenatal diagnosis can be proposed for affected families in which a mutation has been identified. Ursodeoxycholic acid (UDCA) therapy should be initiated in all patients to prevent liver damage. In some PFIC1 or PFIC2 patients, biliary diversion can also relieve pruritus and slow disease progression. However, most PFIC patients are ultimately candidates for liver transplantation. Monitoring of hepatocellular carcinoma, especially in PFIC2 patients, should be offered from the first year of life. Hepatocyte transplantation, gene therapy or specific targeted pharmacotherapy may represent alternative treatments in the future.
Figures

References
-
- Baussan C, Cresteil D, Gonzales E, Raynaud N, Dumont M, Bernard O, Hadchouel M, Jacquemin E. Genetic cholestatic liver diseases:The example of progressive familial intrahepatic cholestasis and related disorders. Acta Gastroenterol Belg. 2004;64:179–183. - PubMed
-
- Bull LN, Carlton VEH, Stricker NL, Baharloo S, DeYoung JA, Freimer NB, Magid MS, Kahn E, Markowitz J, DiCarlo FJ, McLoughlin L, Boyle JT, Dahms BB, Faught PR, Fitzgerald JF, Piccoli DA, Witzleben CL, O'Connell NC, Setchell KD, Agostini RM, Jr, Kocoshis SA, Reyes J, Knisely AS. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): Evidence for heterogeneity. Hepatology. 1997;26:155–164. doi: 10.1002/hep.510260121. - DOI - PubMed
-
- Jansen PLM, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJEJ, Koning JH, de Jager-Krikken A, Kuipers F, Stellard F, Bijleveld CMA, Gouw A, van Goor H, Thompson RJ, Muller M. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/S0016-5085(99)70287-8. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
