

Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A
- PMID: 19136557
- PMCID: PMC2658105
- DOI: 10.1074/jbc.M801892200
Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A
Abstract
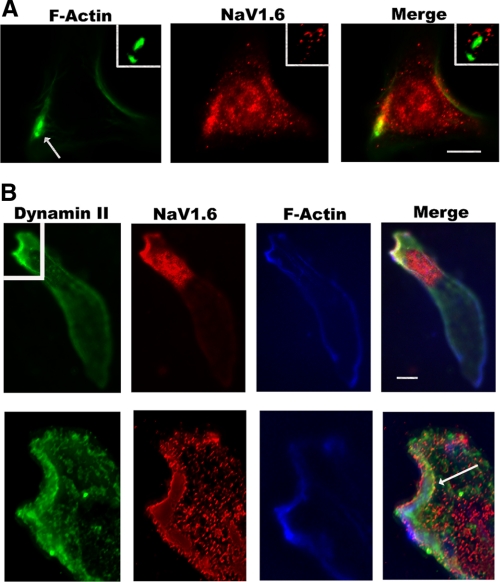
Voltage-gated sodium channels initiate electrical signaling in excitable cells such as muscle and neurons. They also are expressed in non-excitable cells such as macrophages and neoplastic cells. Previously, in macrophages, we demonstrated expression of SCN8A, the gene that encodes the channel NaV1.6, and intracellular localization of NaV1.6 to regions near F-actin bundles, particularly at areas of cell attachment. Here we show that a splice variant of NaV1.6 regulates cellular invasion through its effects on podosome and invadopodia formation in macrophages and melanoma cells. cDNA sequence analysis of SCN8A from THP-1 cells, a human monocyte-macrophage cell line, confirmed the expression of a full-length splice variant that lacks exon 18. Immunoelectron microscopy demonstrated NaV1.6-positive staining within the electron dense podosome rosette structure. Pharmacologic antagonism with tetrodotoxin (TTX) in differentiated THP-1 cells or absence of functional NaV1.6 through a naturally occurring mutation (med) in mouse peritoneal macrophages inhibited podosome formation. Agonist-mediated activation of the channel with veratridine caused release of sodium from cationic vesicular compartments, uptake by mitochondria, and mitochondrial calcium release through the Na/Ca exchanger. Invasion by differentiated THP-1 and HTB-66 cells, an invasive melanoma cell line, through extracellular matrix was inhibited by TTX. THP-1 invasion also was inhibited by small hairpin RNA knockdown of SCN8A. These results demonstrate that a variant of NaV1.6 participates in the control of podosome and invadopodia formation and suggest that intracellular sodium release mediated by NaV1.6 may regulate cellular invasion of macrophages and melanoma cells.
Figures










Similar articles
-
Expression of the voltage-gated sodium channel NaV1.5 in the macrophage late endosome regulates endosomal acidification.J Immunol. 2007 Jun 15;178(12):7822-32. doi: 10.4049/jimmunol.178.12.7822. J Immunol. 2007. PMID: 17548620
-
Electrophysiological and molecular identification of voltage-gated sodium channels in murine vascular myocytes.J Physiol. 2005 Oct 1;568(Pt 1):155-69. doi: 10.1113/jphysiol.2005.090951. Epub 2005 Jul 14. J Physiol. 2005. PMID: 16020462 Free PMC article.
-
Rbfox proteins regulate alternative splicing of neuronal sodium channel SCN8A.Mol Cell Neurosci. 2012 Feb;49(2):120-6. doi: 10.1016/j.mcn.2011.10.005. Epub 2011 Oct 21. Mol Cell Neurosci. 2012. PMID: 22044765 Free PMC article.
-
Sodium Channelopathies in Human and Animal Models of Epilepsy and Neurodevelopmental Disorders.In: Noebels JL, Avoli M, Rogawski MA, Vezzani A, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 5th edition. New York: Oxford University Press; 2024. Chapter 44. In: Noebels JL, Avoli M, Rogawski MA, Vezzani A, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 5th edition. New York: Oxford University Press; 2024. Chapter 44. PMID: 39637100 Free Books & Documents. Review.
-
Cerebellum-related characteristics of Scn8a-mutant mice.Cerebellum. 2009 Sep;8(3):192-201. doi: 10.1007/s12311-009-0110-z. Epub 2009 May 8. Cerebellum. 2009. PMID: 19424768 Review.
Cited by
-
Voltage-gated sodium channels in cancers.Biomark Res. 2024 Jul 25;12(1):70. doi: 10.1186/s40364-024-00620-x. Biomark Res. 2024. PMID: 39060933 Free PMC article. Review.
-
Endoplasmic reticulum export, subcellular distribution, and fibril formation by Pmel17 require an intact N-terminal domain junction.J Biol Chem. 2010 May 21;285(21):16166-83. doi: 10.1074/jbc.M109.097725. Epub 2010 Mar 15. J Biol Chem. 2010. PMID: 20231267 Free PMC article.
-
Voltage-gated sodium channels and cancer: is excitability their primary role?Front Pharmacol. 2015 Jul 29;6:152. doi: 10.3389/fphar.2015.00152. eCollection 2015. Front Pharmacol. 2015. PMID: 26283962 Free PMC article. Review.
-
Bioelectric Control of Metastasis in Solid Tumors.Bioelectricity. 2019 Sep 1;1(3):114-130. doi: 10.1089/bioe.2019.0013. Epub 2019 Sep 16. Bioelectricity. 2019. PMID: 32292893 Free PMC article.
-
Sodium channels and pain: from toxins to therapies.Br J Pharmacol. 2018 Jun;175(12):2138-2157. doi: 10.1111/bph.13962. Epub 2017 Sep 2. Br J Pharmacol. 2018. PMID: 28749537 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
