

DNA repair deficiency and neurological disease
- PMID: 19145234
- PMCID: PMC3064843
- DOI: 10.1038/nrn2559
DNA repair deficiency and neurological disease
Erratum in
- Nat Rev Neurosci. 2009 Mar;10(3):242
Abstract
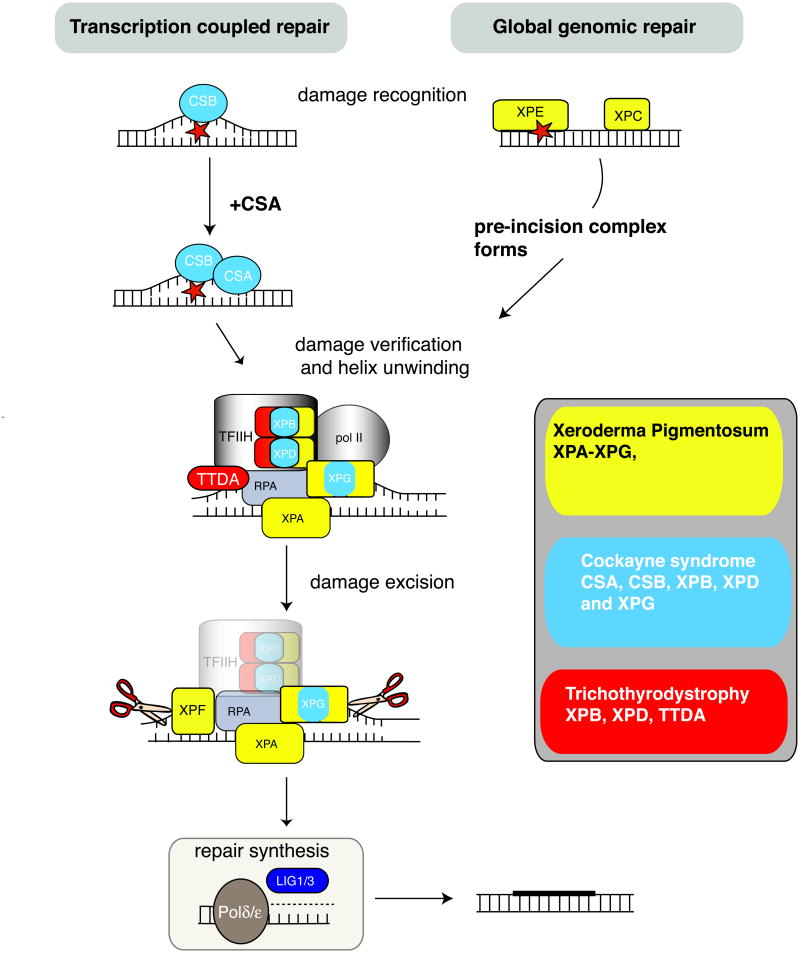
The ability to respond to genotoxic stress is a prerequisite for the successful development of the nervous system. Mutations in various DNA repair factors can lead to human diseases that are characterized by pronounced neuropathology. In many of these syndromes the neurological component is among the most deleterious aspects of the disease. The nervous system poses a particular challenge in terms of clinical intervention, as the neuropathology associated with these diseases often arises during nervous system development and can be fully penetrant by childhood. Understanding how DNA repair deficiency affects the nervous system will provide a rational basis for therapies targeted at ameliorating the neurological problems in these syndromes.
Figures
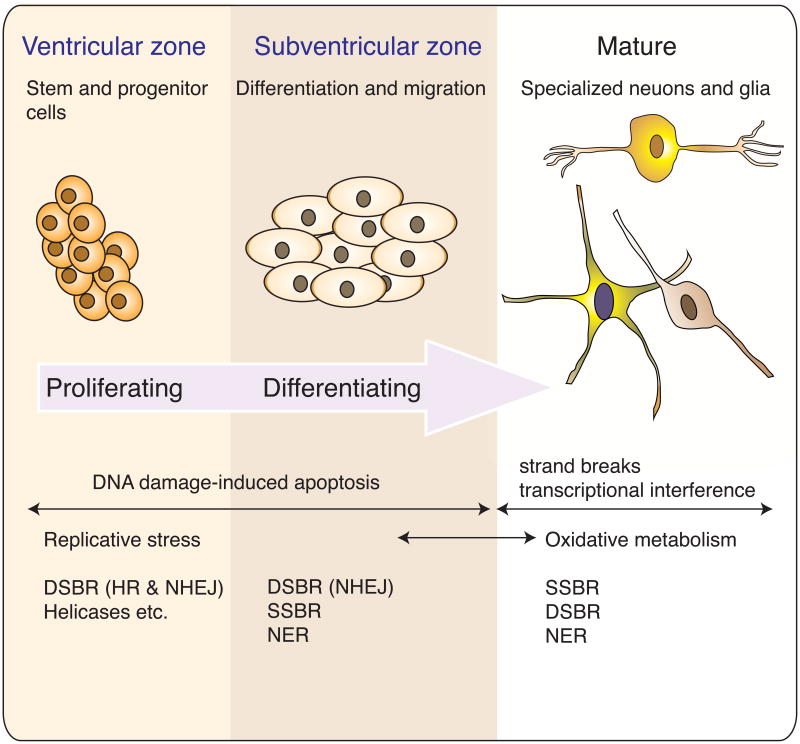
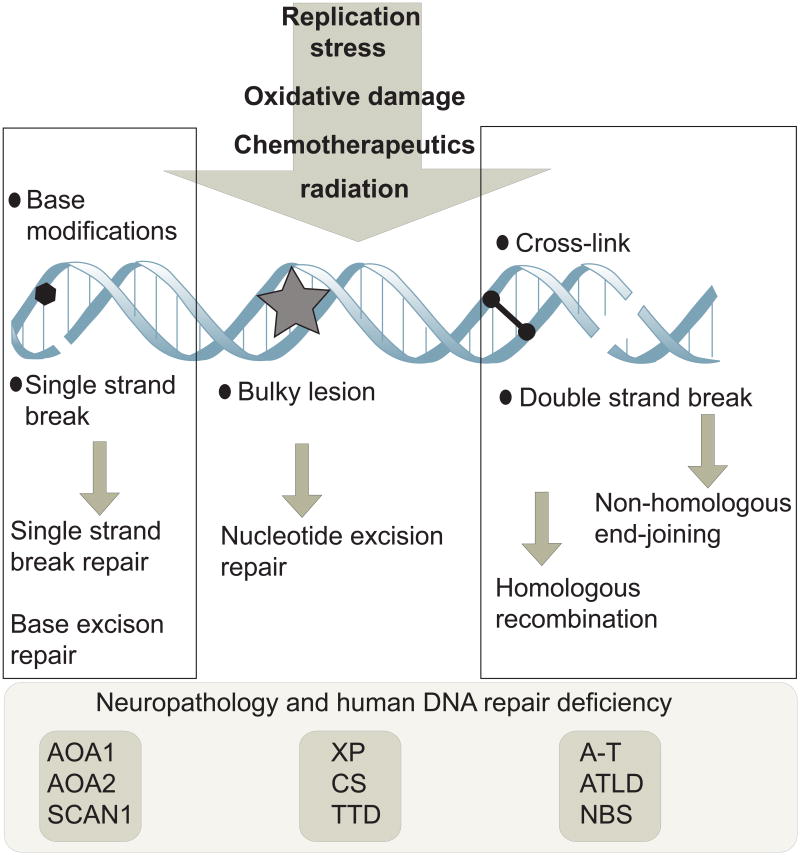
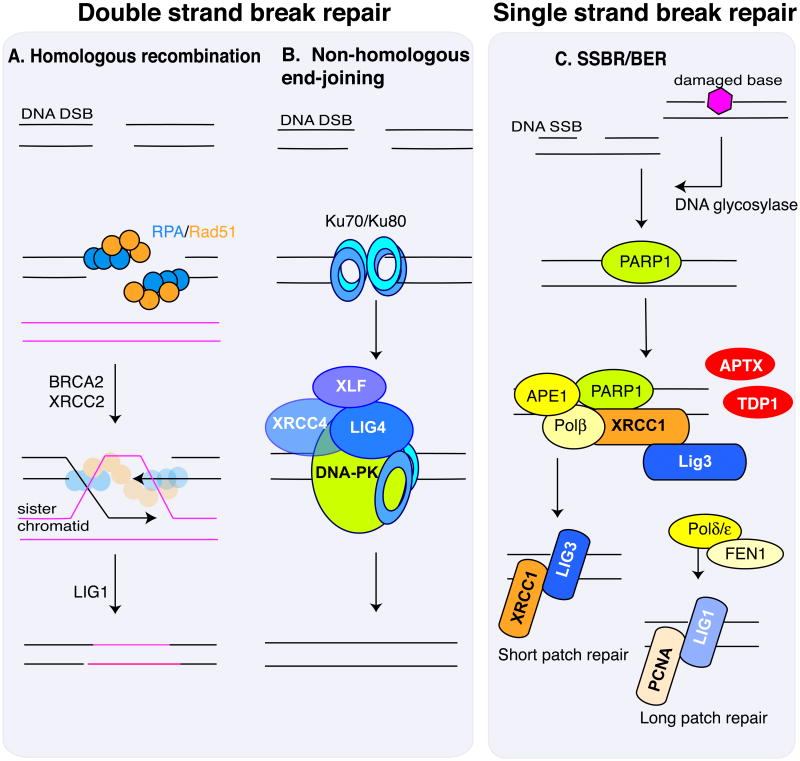
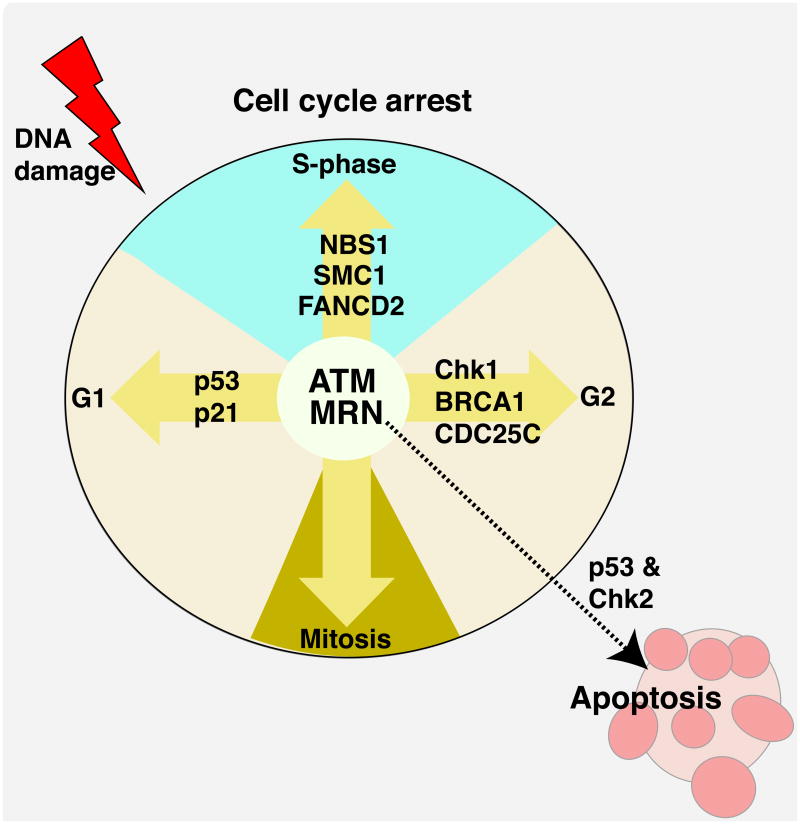
References
-
- Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7. - PubMed
-
- Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–20. - PubMed
-
- Nouspikel T, Hanawalt PC. When parsimony backfires: neglecting DNA repair may doom neurons in Alzheimer's disease. Bioessays. 2003;25:168–73. - PubMed
-
- Chan WY, Lorke DE, Tiu SC, Yew DT. Proliferation and apoptosis in the developing human neocortex. Anat Rec. 2002;267:261–76. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
