

Computations of standard binding free energies with molecular dynamics simulations
- PMID: 19146384
- PMCID: PMC3837708
- DOI: 10.1021/jp807701h
Computations of standard binding free energies with molecular dynamics simulations
Abstract
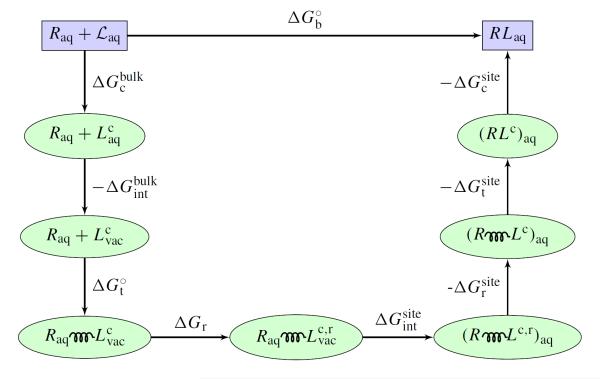
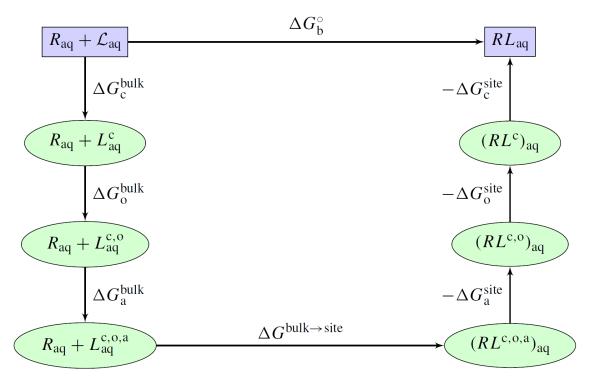
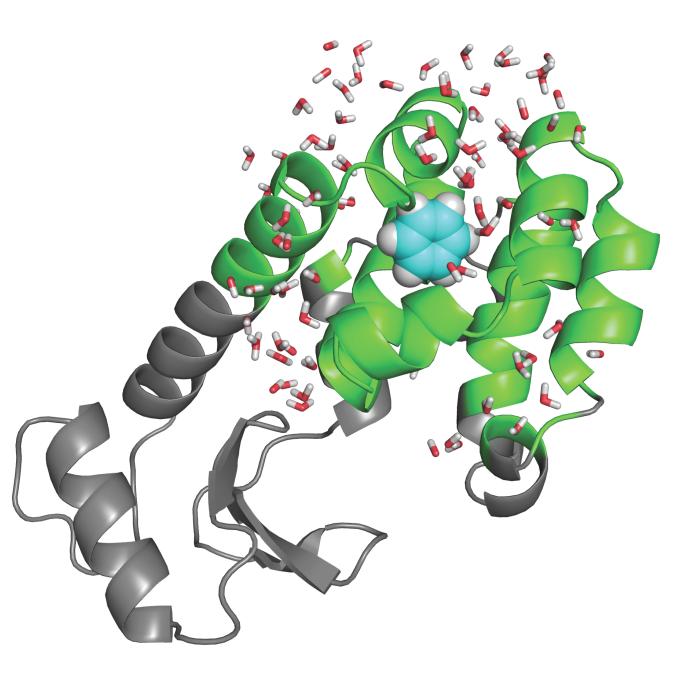
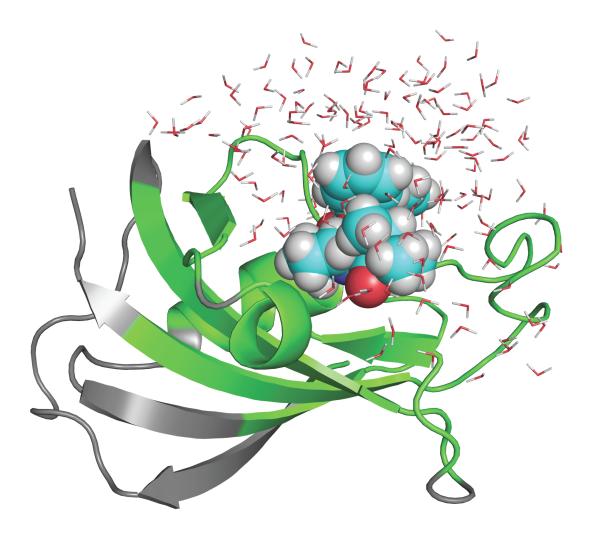
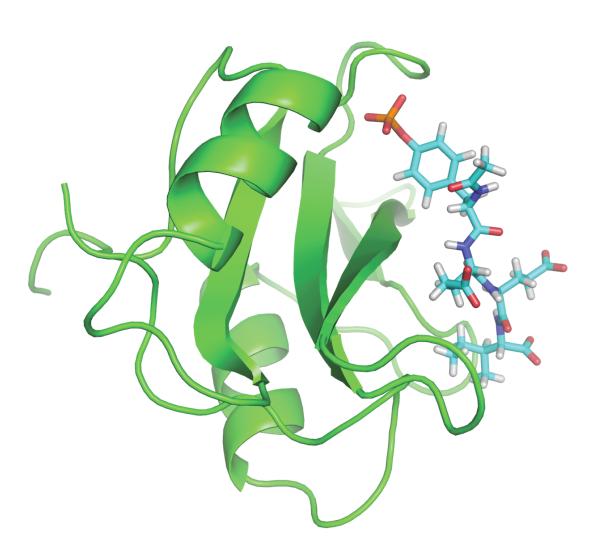
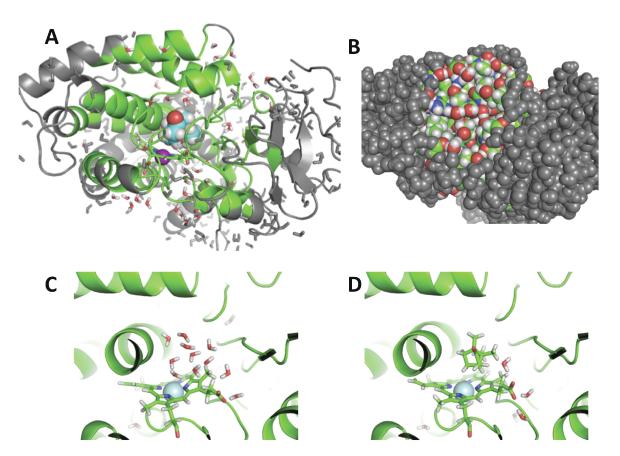
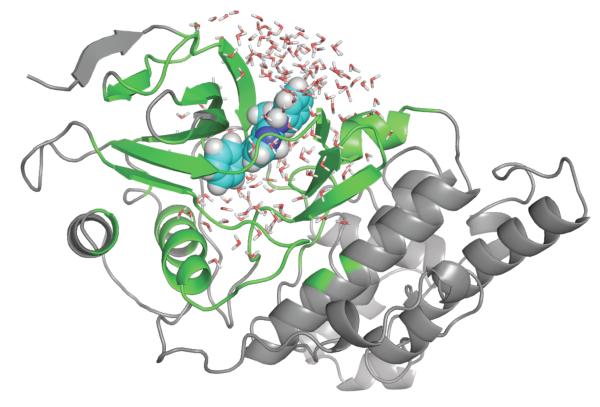
An increasing number of studies have reported computations of the standard (absolute) binding free energy of small ligands to proteins using molecular dynamics (MD) simulations and explicit solvent molecules that are in good agreement with experiments. This encouraging progress suggests that physics-based approaches hold the promise of making important contributions to the process of drug discovery and optimization in the near future. Two types of approaches are principally used to compute binding free energies with MD simulations. The most widely known is the alchemical double decoupling method, in which the interaction of the ligand with its surroundings are progressively switched off. It is also possible to use a potential of mean force (PMF) method, in which the ligand is physically separated from the protein receptor. For both of these computational approaches, restraining potentials may be activated and released during the simulation for sampling efficiently the changes in translational, rotational, and conformational freedom of the ligand and protein upon binding. Because such restraining potentials add bias to the simulations, it is important that their effects be rigorously removed to yield a binding free energy that is properly unbiased with respect to the standard state. A review of recent results is presented, and differences in computational methods are discussed. Examples of computations with T4-lysozyme mutants, FKBP12, SH2 domain, and cytochrome P450 are discussed and compared. Remaining difficulties and challenges are highlighted.
Figures
References
-
- Vindigni A. Energetic dissection of specificity in serine proteases. Comb. Chem. High Throughput Screen. 1999;2:139–153. - PubMed
-
- Cheng AC, Calabro V, Frankel AD. Design of rna-binding proteins and ligands. Curr. Opin. Struct. Biol. 2001;11(4):478–484. - PubMed
-
- Garvie CW, Wolberger C. Recognition of specific dna sequences. Mol. Cell. 2001;8(5):937–946. - PubMed
-
- Shoichet B, Leach A, Kuntz I. Ligand solvation in molecular docking. Proteins. 1999;34:4–16. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
