

Stochastic gating and drug-ribosome interactions
- PMID: 19146858
- PMCID: PMC5970275
- DOI: 10.1016/j.jmb.2008.12.035
Stochastic gating and drug-ribosome interactions
Abstract
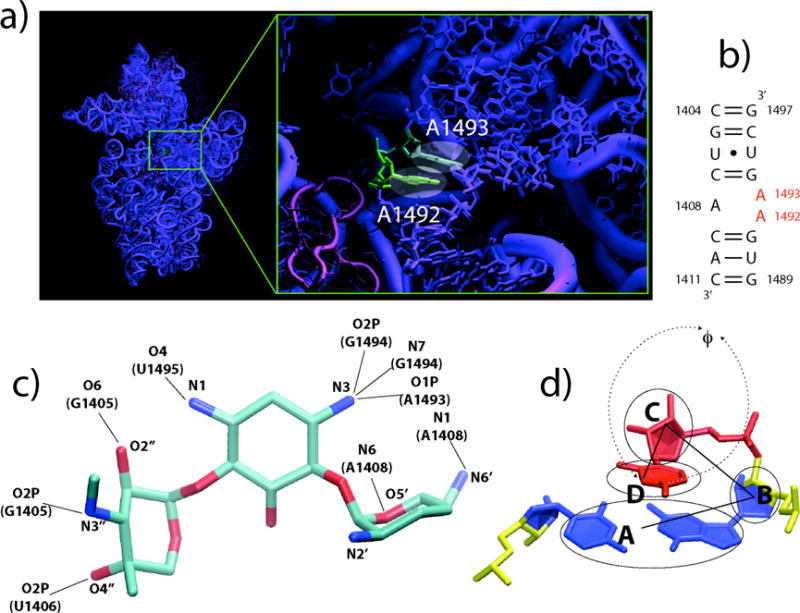
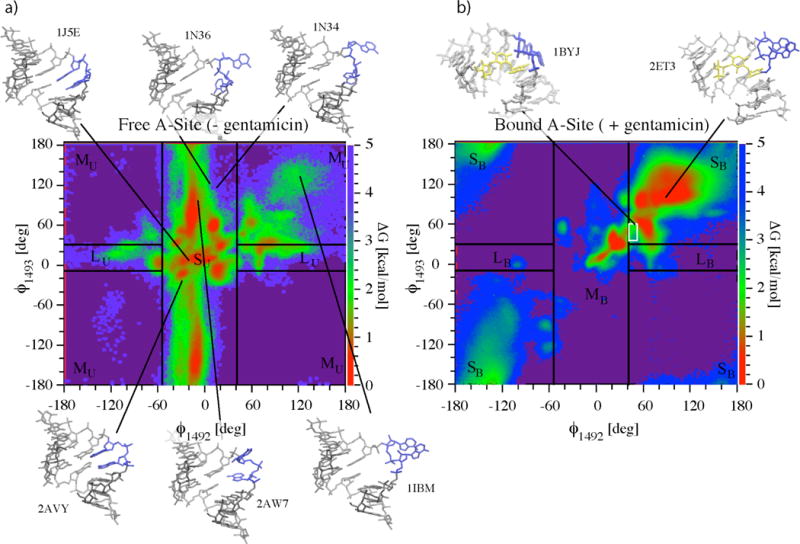
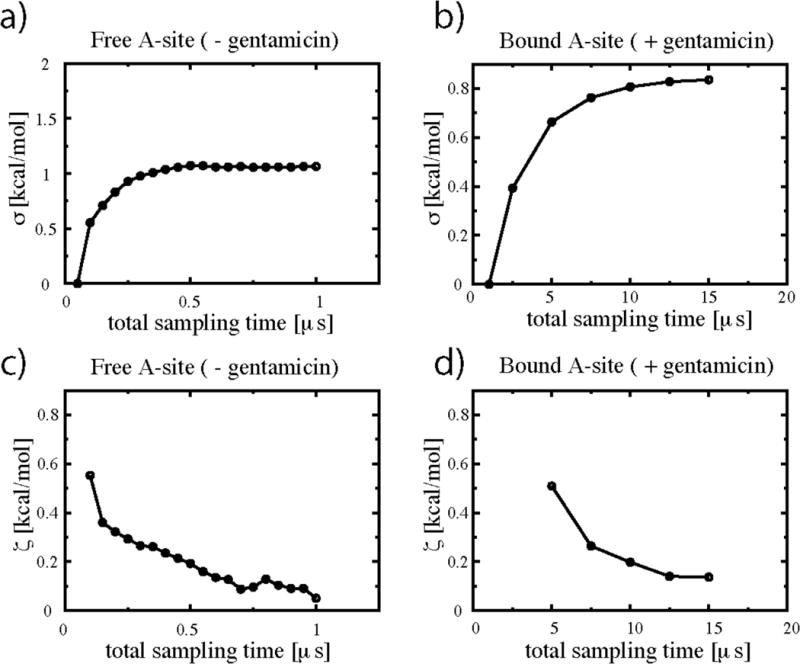
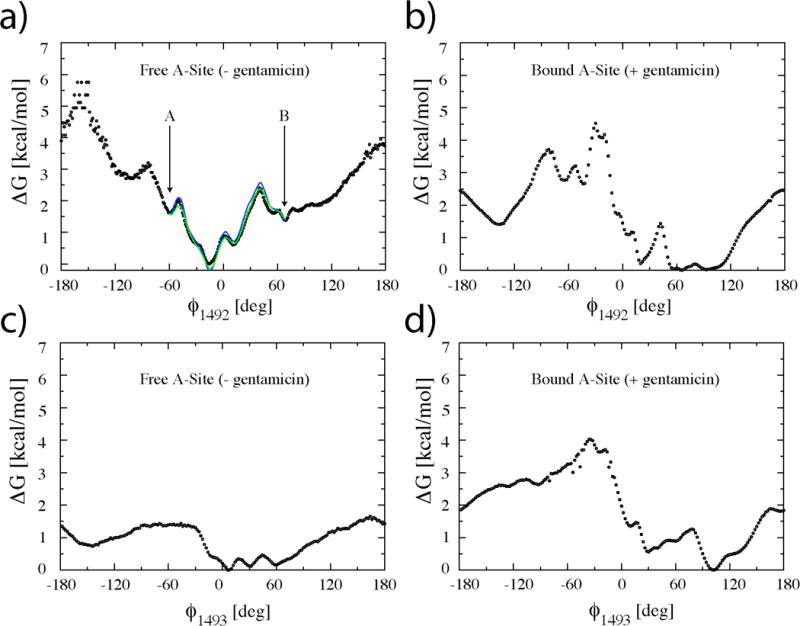
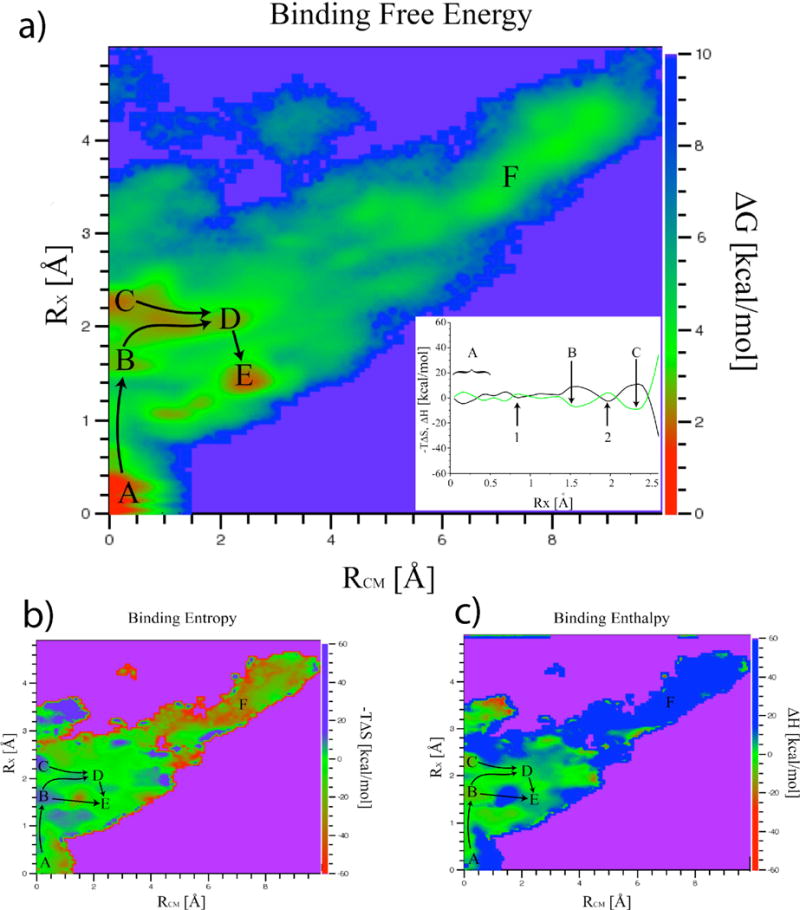
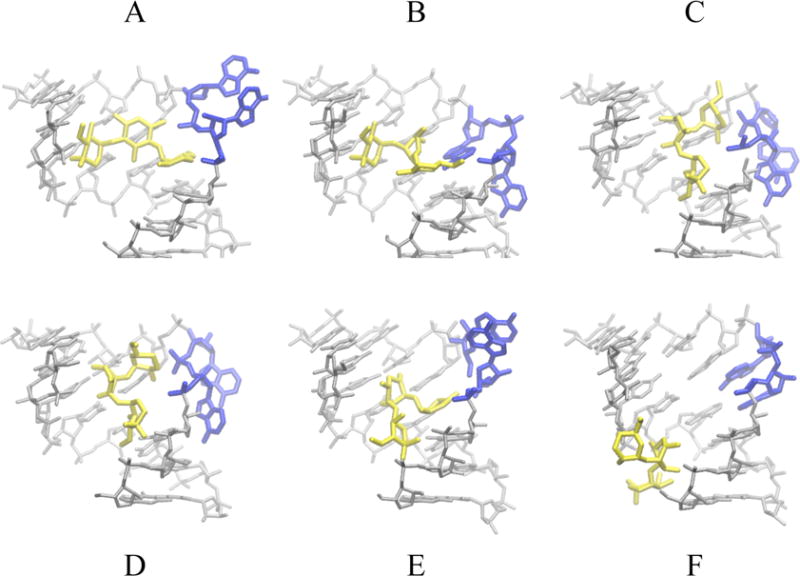
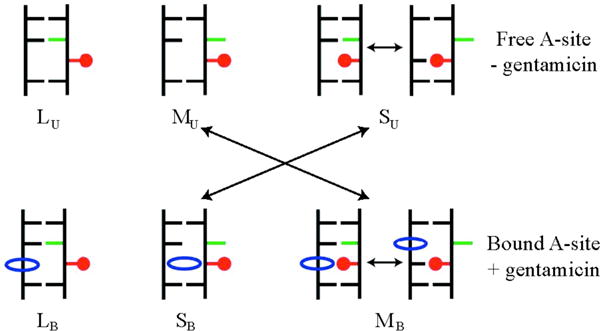
Gentamicin is a potent antibiotic that is used in combination therapy for inhalation anthrax disease. The drug is also often used in therapy for methicillin-resistant Staphylococcusaureus. Gentamicin works by flipping a conformational switch on the ribosome, disrupting the reading head (i.e., 16S ribosomal decoding bases 1492-1493) used for decoding messenger RNA. We use explicit solvent all-atom molecular simulation to study the thermodynamics of the ribosomal decoding site and its interaction with gentamicin. The replica exchange molecular dynamics simulations used an aggregate sampling of 15 mus when summed over all replicas, allowing us to explicitly calculate the free-energy landscape, including a rigorous treatment of enthalpic and entropic effects. Here, we show that the decoding bases flip on a timescale faster than that of gentamicin binding, supporting a stochastic gating mechanism for antibiotic binding, rather than an induced-fit model where the bases only flip in the presence of a ligand. The study also allows us to explore the nonspecific binding landscape near the binding site and reveals that, rather than a two-state bound/unbound scenario, drug dissociation entails shuttling between many metastable local minima in the free-energy landscape. Special care is dedicated to validation of the obtained results, both by direct comparison to experiment and by estimation of simulation convergence.
Figures
References
-
- Mankin A. Nature structural & molecular biology. 2006;13:858–860. - PubMed
-
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340–8. - PubMed
-
- Wimberly BT, Brodersen DE, Clemons WM, Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Nature. 2000;407:327–39. - PubMed
-
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Science (New York, NY. 2005;310:827–34. - PubMed
-
- Korostelev A, Trakhanov S, Laurberg M, Noller HF. Cell. 2006;126:1065–77. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
