

Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition
- PMID: 19147570
- PMCID: PMC2737189
- DOI: 10.1158/0008-5472.CAN-08-3389
Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition
Abstract
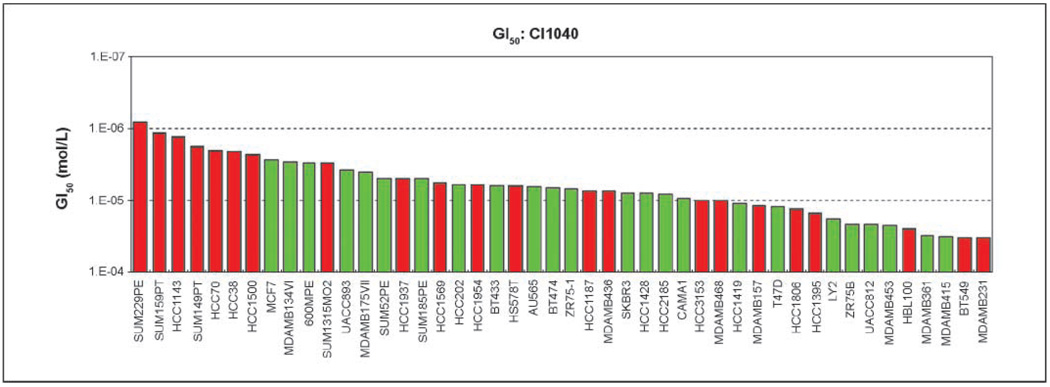
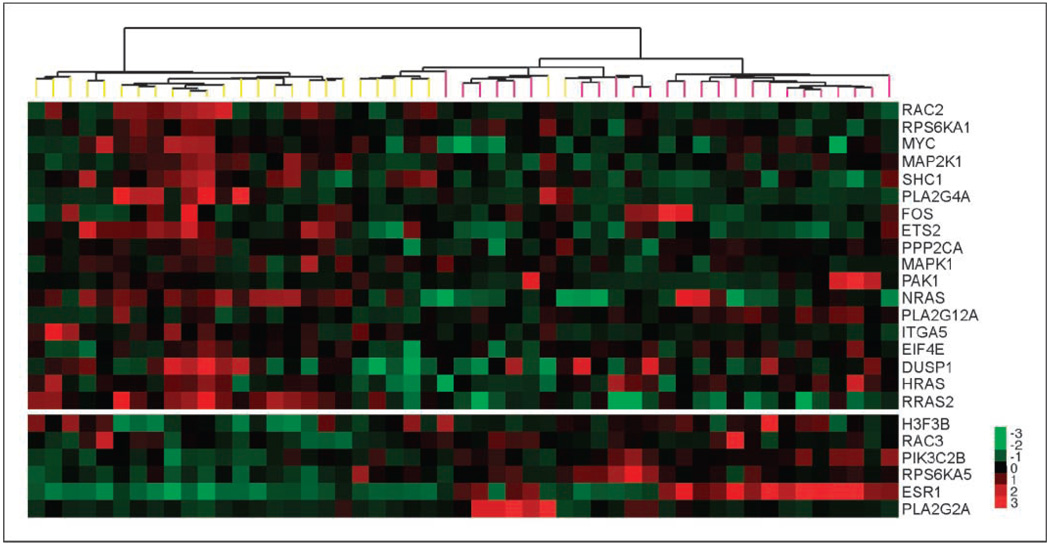
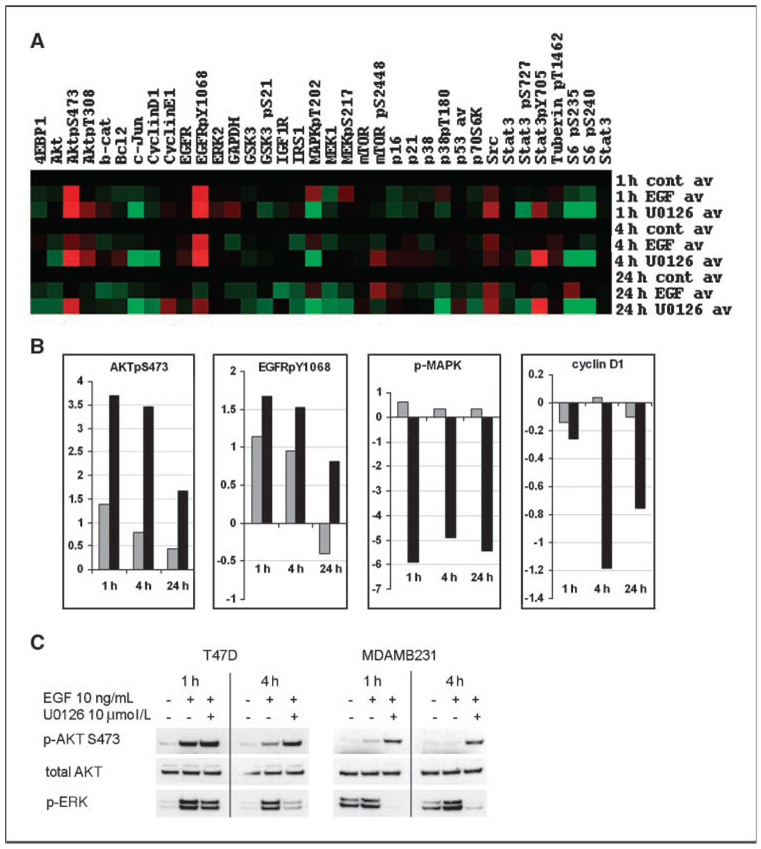
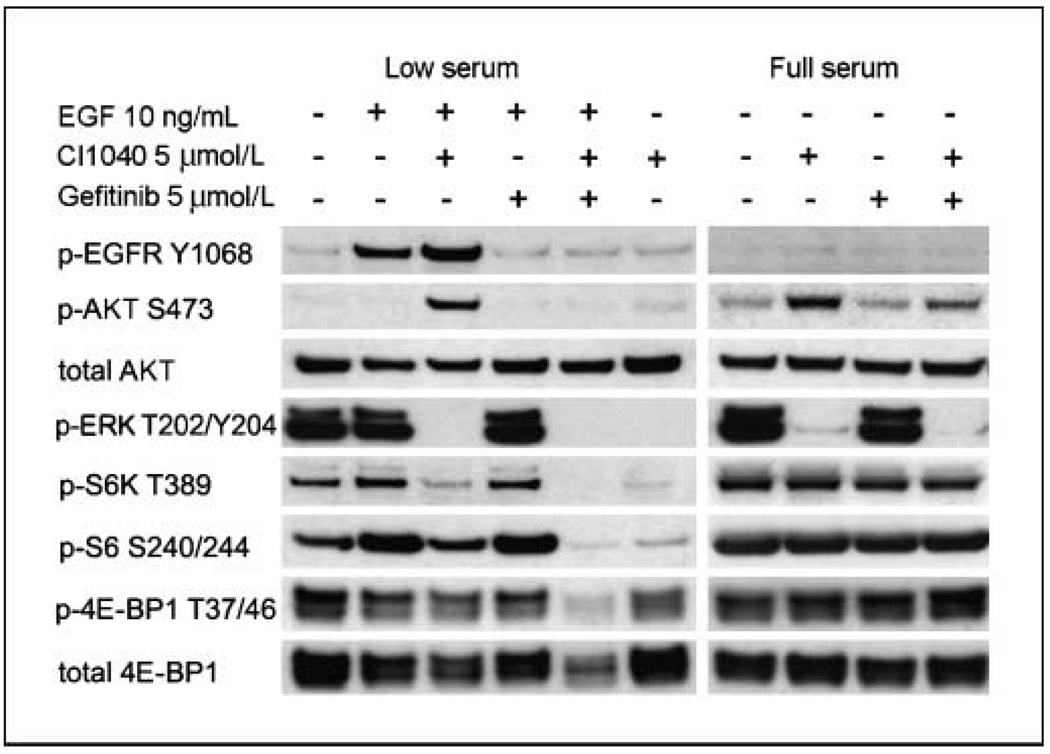
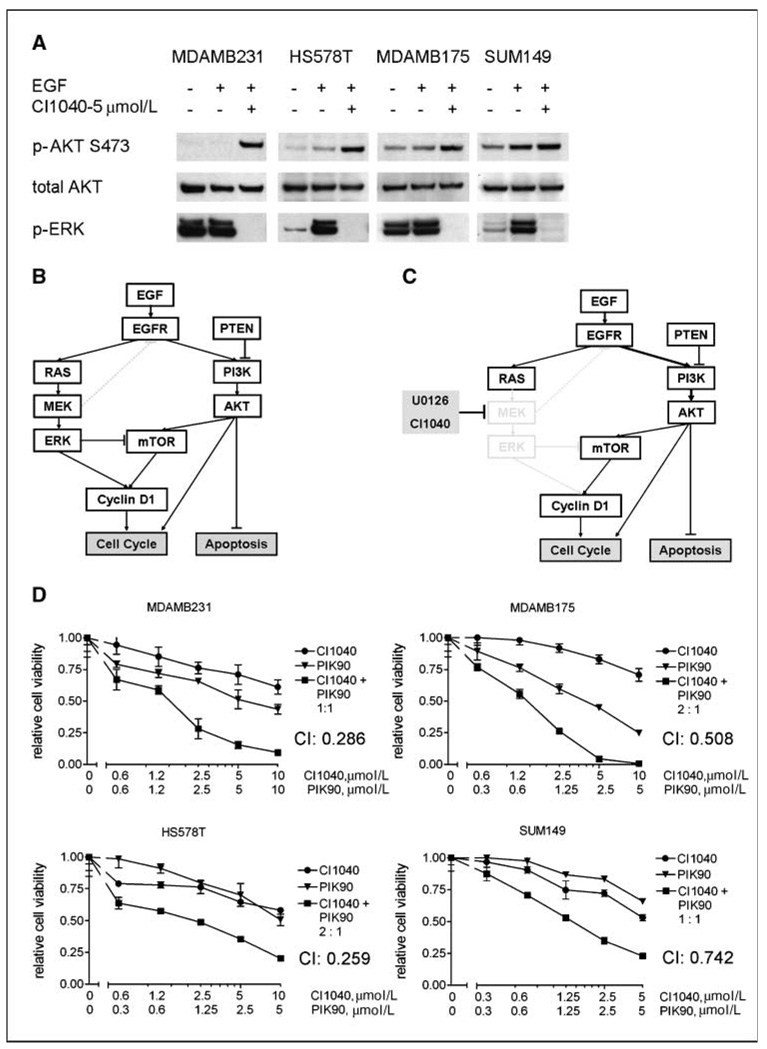
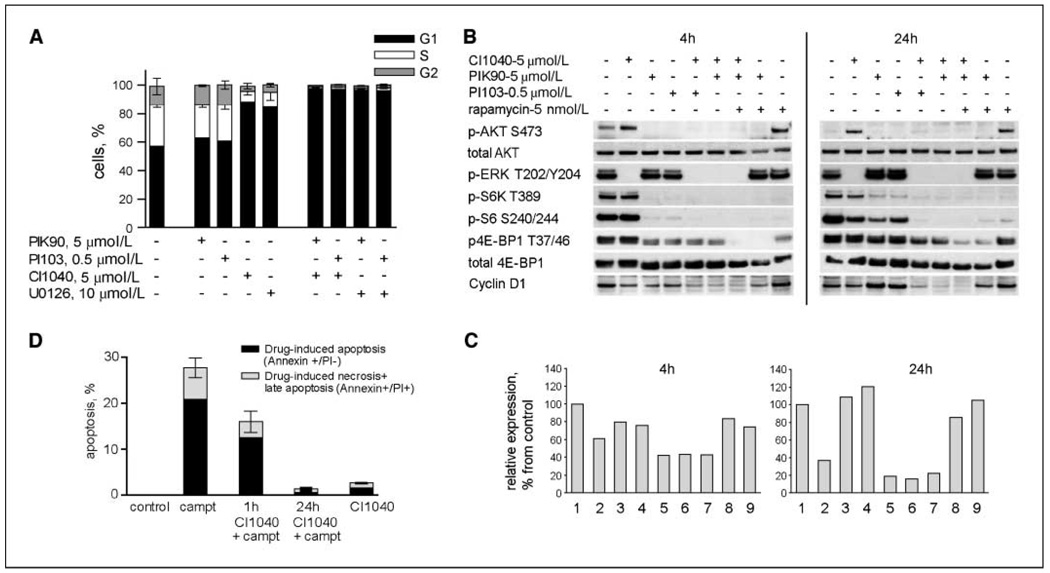
Specific inhibitors of mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK) have been developed that efficiently inhibit the oncogenic RAF-MEK-ERK pathway. We used a systems-based approach to identify breast cancer subtypes particularly susceptible to MEK inhibitors and to understand molecular mechanisms conferring resistance to such compounds. Basal-type breast cancer cells were found to be particularly susceptible to growth inhibition by small-molecule MEK inhibitors. Activation of the phosphatidylinositol 3-kinase (PI3K) pathway in response to MEK inhibition through a negative MEK-epidermal growth factor receptor-PI3K feedback loop was found to limit efficacy. Interruption of this feedback mechanism by targeting MEK and PI3K produced synergistic effects, including induction of apoptosis and, in some cell lines, cell cycle arrest and protection from apoptosis induced by proapoptotic agents. These findings enhance our understanding of the interconnectivity of oncogenic signal transduction circuits and have implications for the design of future clinical trials of MEK inhibitors in breast cancer by guiding patient selection and suggesting rational combination therapies.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures
References
-
- Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–2559. - PubMed
-
- Perez-Tenorio G, Alkhori L, Olsson B, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. 2007;13:3577–3584. - PubMed
-
- Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. - PubMed
-
- Rochlitz CF, Scott GK, Dodson JM, et al. Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res. 1989;49:357–360. - PubMed
-
- Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12:104–107. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
