

Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo
- PMID: 19147578
- PMCID: PMC3418672
- DOI: 10.1158/0008-5472.CAN-08-1191
Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo
Erratum in
- Cancer Res. 2013 Jul 1;73(13):4170
Abstract
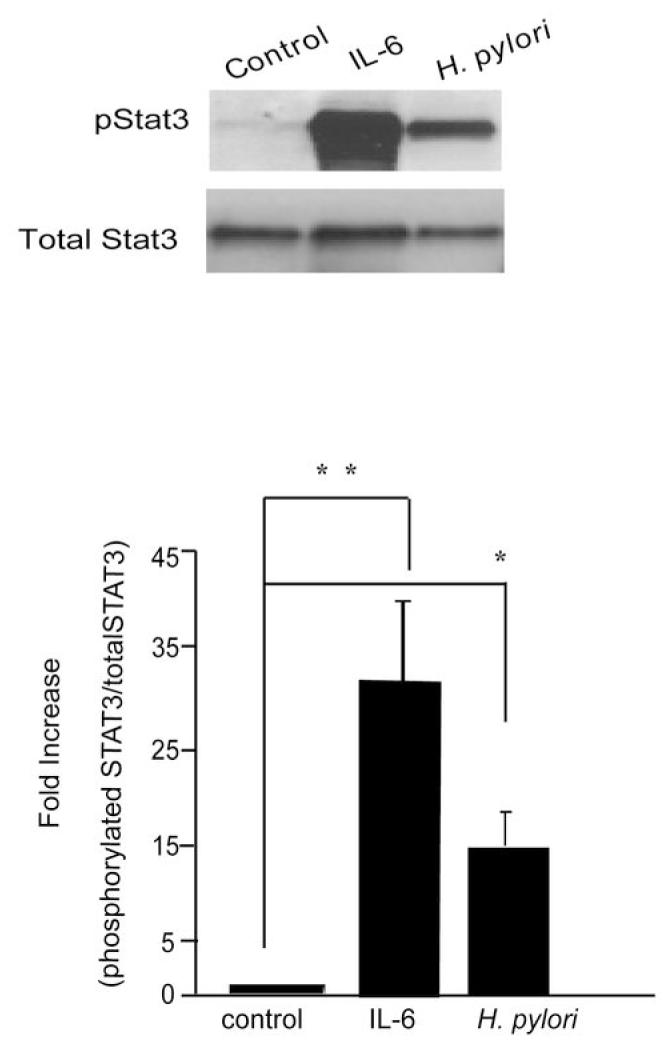
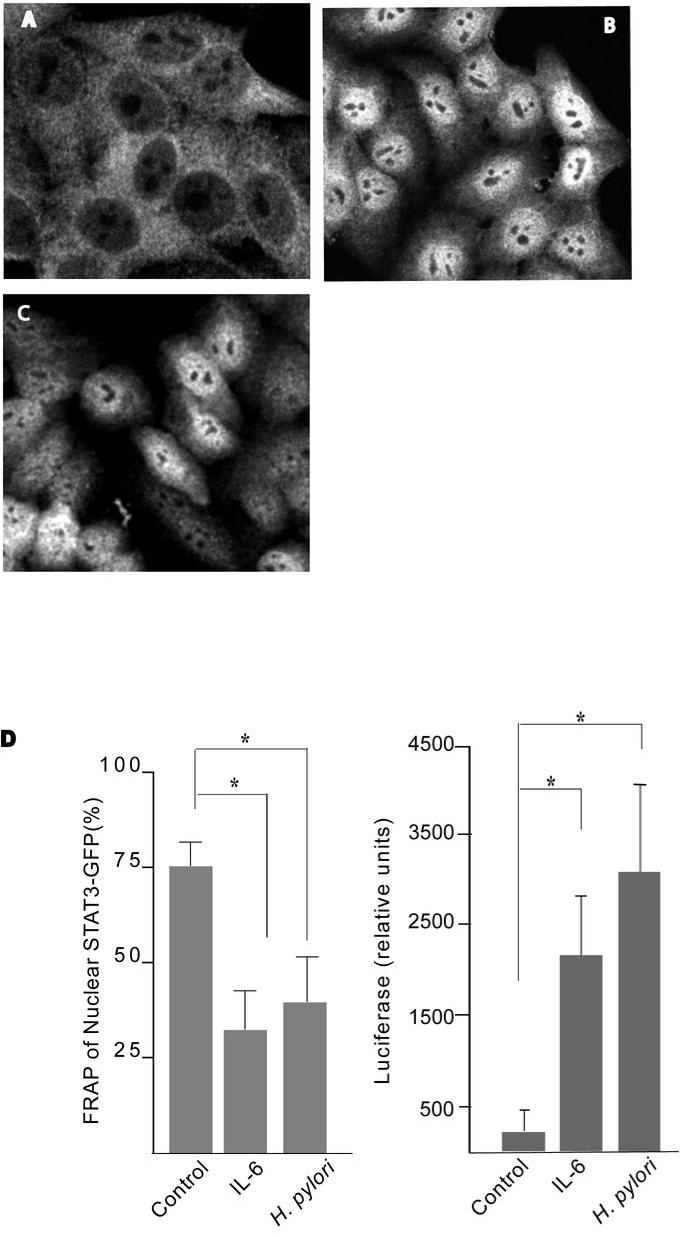
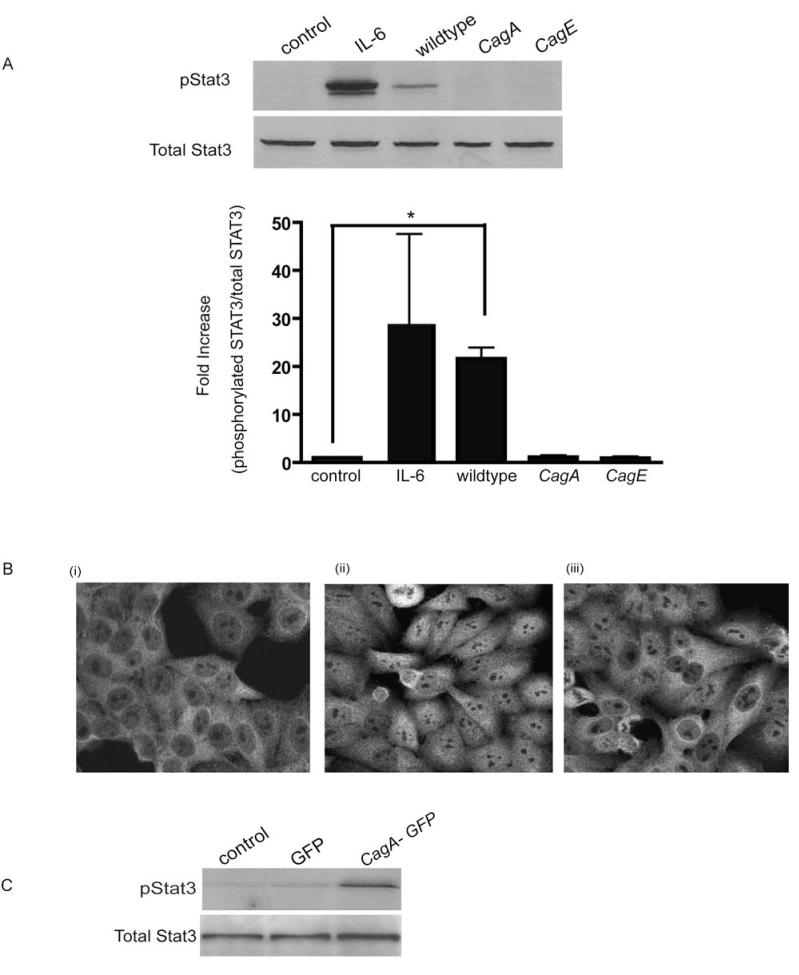
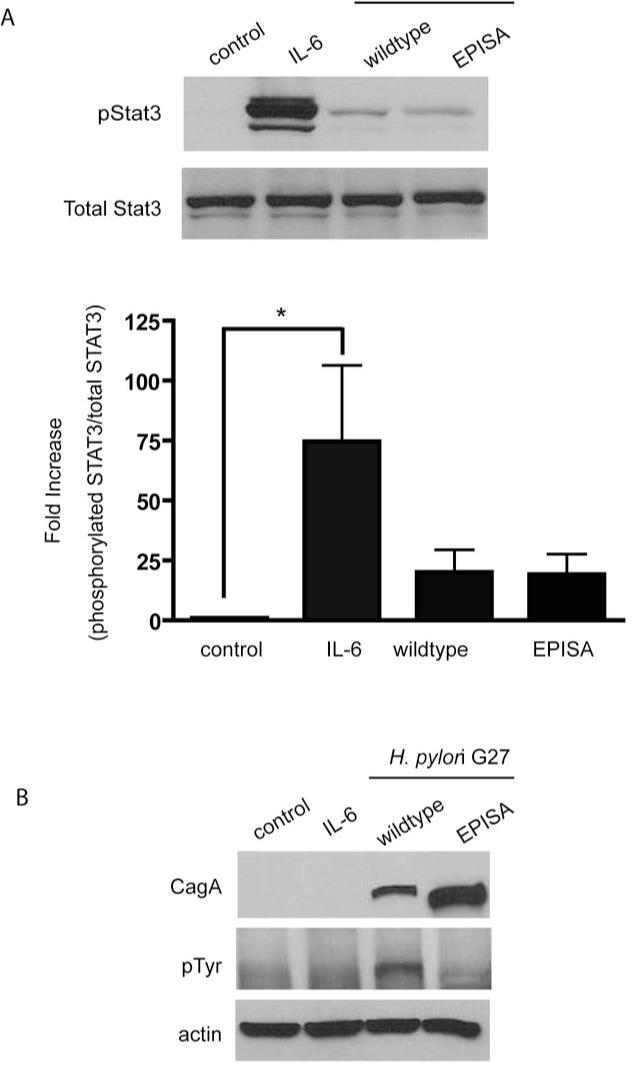
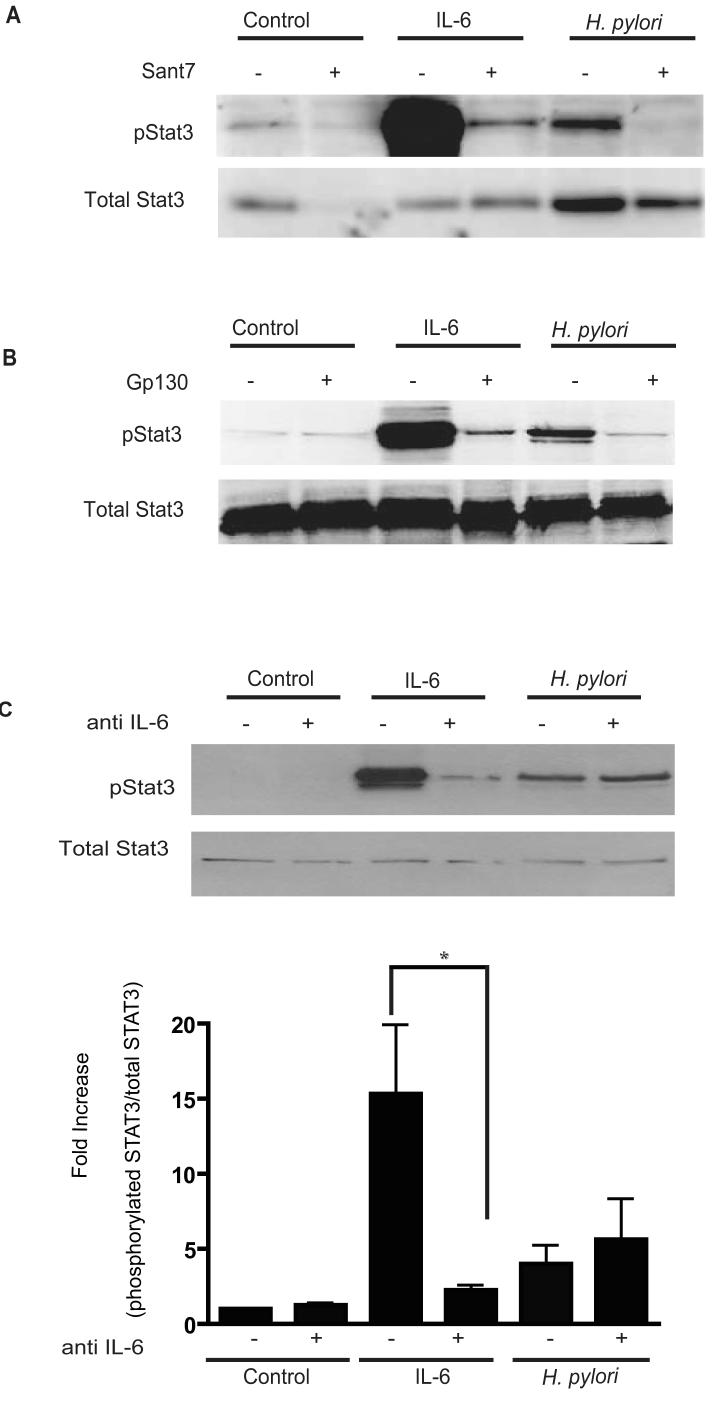
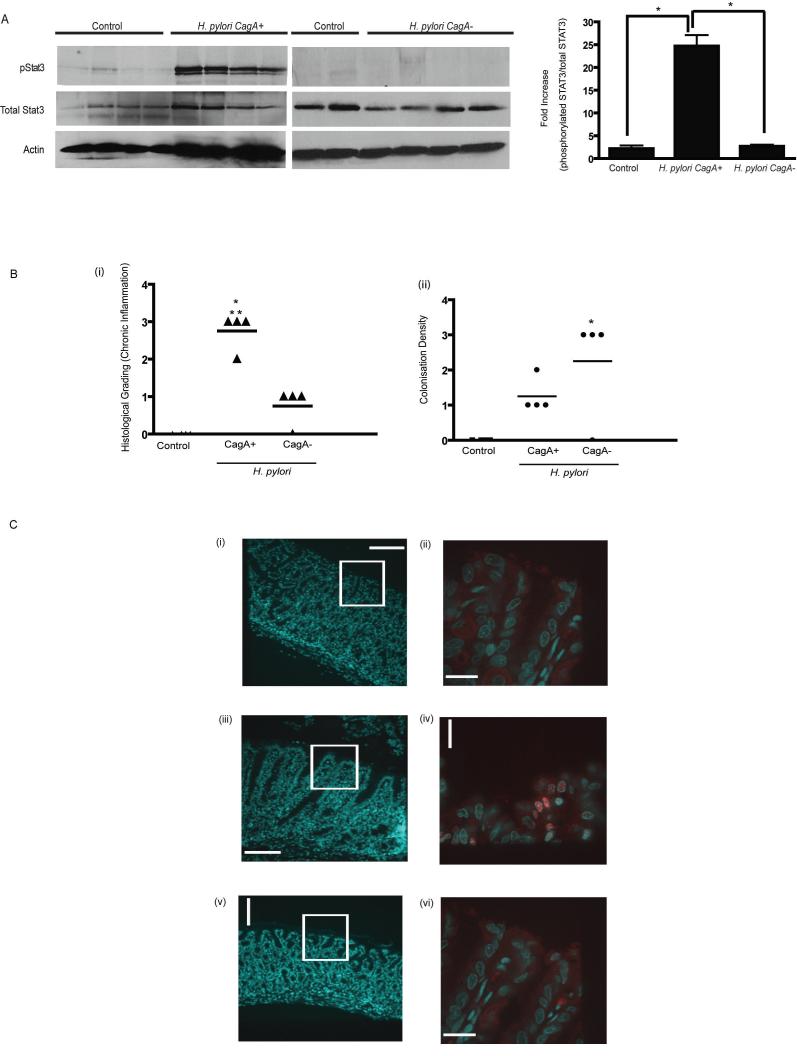
Persistent infection with Helicobacter pylori confers an increased risk for the development of gastric cancer. However, the exact mechanisms whereby this bacterium causes carcinogenesis have not been completely elucidated. Recent evidence indicates that aberrant activation of the signal transducers and activators of transcription 3 (STAT3) signaling pathway may play a role in gastric carcinogenesis. Therefore, we hypothesized that H. pylori infection modulates STAT3 signaling, favoring gastric cancer development. In epithelial cells infected with H. pylori, STAT3 was activated, as assessed by immunoblotting for phosphorylated STAT3, immunofluorescence of translocated STAT3, fluorescence recovery after photobleaching, and luciferase activation in transfected cells. Activation was dependent on translocation but not phosphorylation of cytotoxin-associated gene A (CagA) in host cells. Activation seemed to be receptor-mediated because preincubation of cells with the interleukin-6 (IL-6) receptor superantagonist sant7 or inhibition of gp130 by a monoclonal antibody prevented H. pylori-mediated STAT3 activation. However, activation was not related to autocrine activation by IL-6 or IL-11. CagA+ wild-type H. pylori, but not the noncarcinogenic cagA- mutant, activated STAT3 in gastric epithelial cells in vivo in the gerbil model of H. pylori-mediated gastric carcinogenesis. Collectively, these results indicate that H. pylori CagA activates the STAT3 signaling pathway in vitro and in vivo, providing a potential mechanism by which chronic H. pylori infection promotes the development of gastric cancer.
Figures
References
-
- Peek RM, Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–48. - PubMed
-
- Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–94. - PubMed
-
- Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2000;10:745–55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
