

Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice
- PMID: 19150977
- PMCID: PMC2721639
- DOI: 10.1093/cvr/cvp016
Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice
Abstract
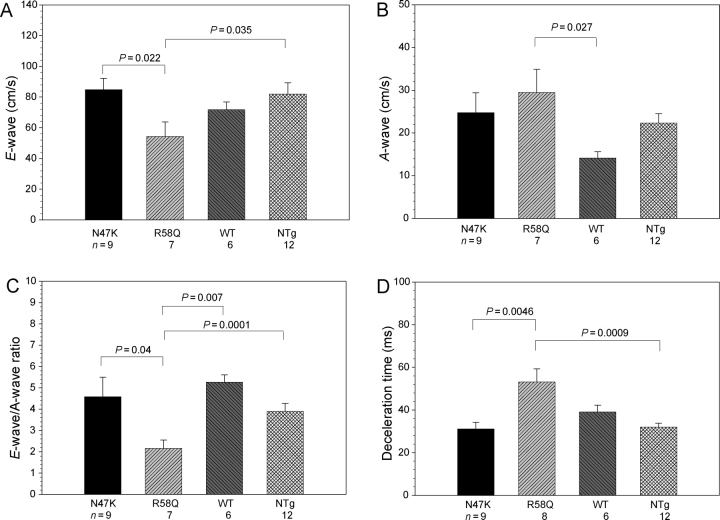
Aims: Several mutations in the ventricular myosin regulatory light chain (RLC) were identified to cause familial hypertrophic cardiomyopathy (FHC). Based on our previous cellular findings showing delayed calcium transients in electrically stimulated intact papillary muscle fibres from transgenic Tg-R58Q and Tg-N47K mice and, in addition, prolonged force transients in Tg-R58Q fibres, we hypothesized that the malignant FHC phenotype associated with the R58Q mutation is most likely related to diastolic dysfunction.
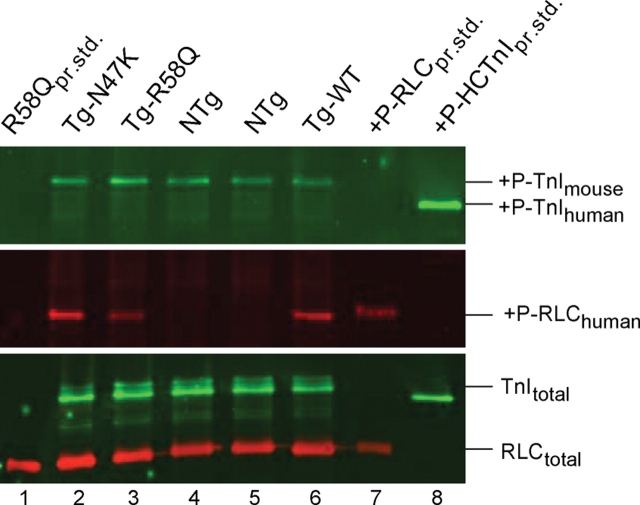
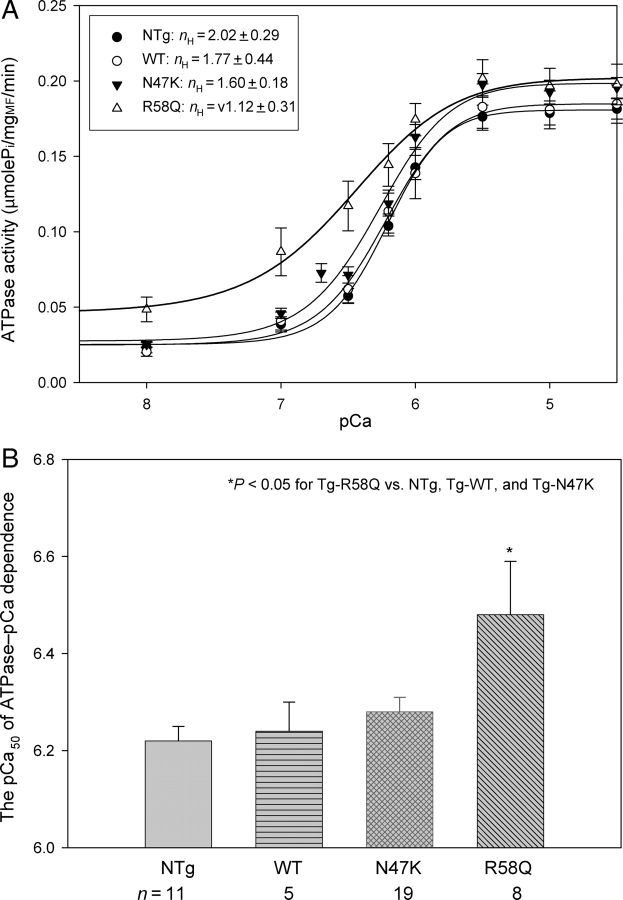
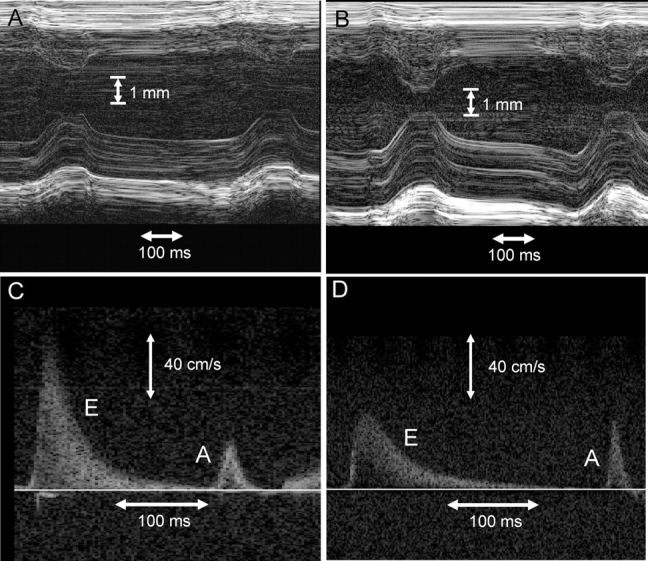
Methods and results: Cardiac morphology and in vivo haemodynamics by echocardiography as well as cardiac function in isolated perfused working hearts were assessed in transgenic (Tg) mutant mice. The ATPase-pCa relationship was determined in myofibrils isolated from Tg mouse hearts. In addition, the effect of both mutations on RLC phosphorylation was examined in rapidly frozen ventricular samples from Tg mice. Significantly, decreased cardiac function was observed in isolated perfused working hearts from both Tg-R58Q and Tg-N47K mice. However, echocardiographic examination showed significant alterations in diastolic transmitral velocities and deceleration time only in Tg-R58Q myocardium. Likewise, changes in Ca(2+) sensitivity, cooperativity, and an elevated level of ATPase activity at low [Ca(2+)] were only observed in myofibrils from Tg-R58Q mice. In addition, the R58Q mutation and not the N47K led to reduced RLC phosphorylation in Tg ventricles.
Conclusion: Our results suggest that the N47K and R58Q mutations may act through similar mechanisms, leading to compensatory hypertrophy of the functionally compromised myocardium, but the malignant R58Q phenotype is most likely associated with more severe alterations in cardiac performance manifested as impaired relaxation and global diastolic dysfunction. At the molecular level, we suggest that by reducing the phosphorylation of RLC, the R58Q mutation decreases the kinetics of myosin cross-bridges, leading to an increased myofilament calcium sensitivity and to overall changes in intracellular Ca(2+) homeostasis.
Figures

References
-
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. - PubMed
-
- Seidman CE, Seidman JG. Molecular genetic studies of familial hypertrophic cardiomyopathy. Basic Res Cardiol. 1998;93:13–16. - PubMed
-
- Tardiff J. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. - PubMed
-
- Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19:104–110. - PubMed
-
- Szczesna D. Regulatory light chains of striated muscle myosin. Structure, function and malfunction. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:187–197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
