

A non-Smad mechanism of fibroblast activation by transforming growth factor-beta via c-Abl and Egr-1: selective modulation by imatinib mesylate
- PMID: 19151753
- PMCID: PMC4006376
- DOI: 10.1038/onc.2008.479
A non-Smad mechanism of fibroblast activation by transforming growth factor-beta via c-Abl and Egr-1: selective modulation by imatinib mesylate
Abstract
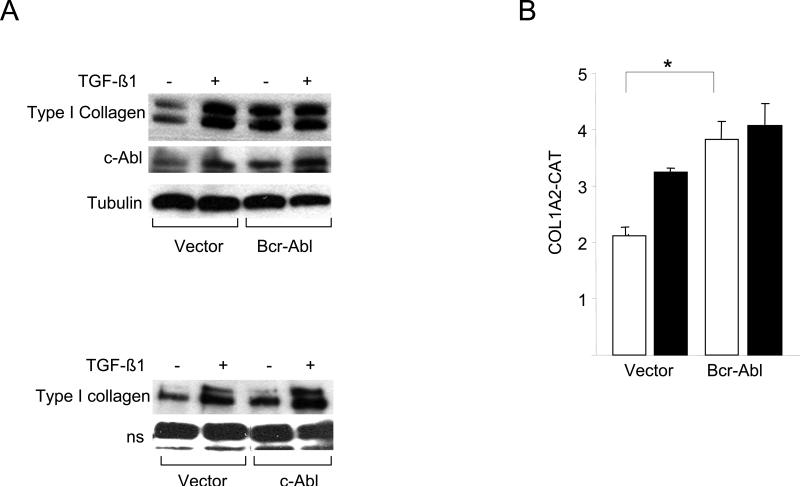
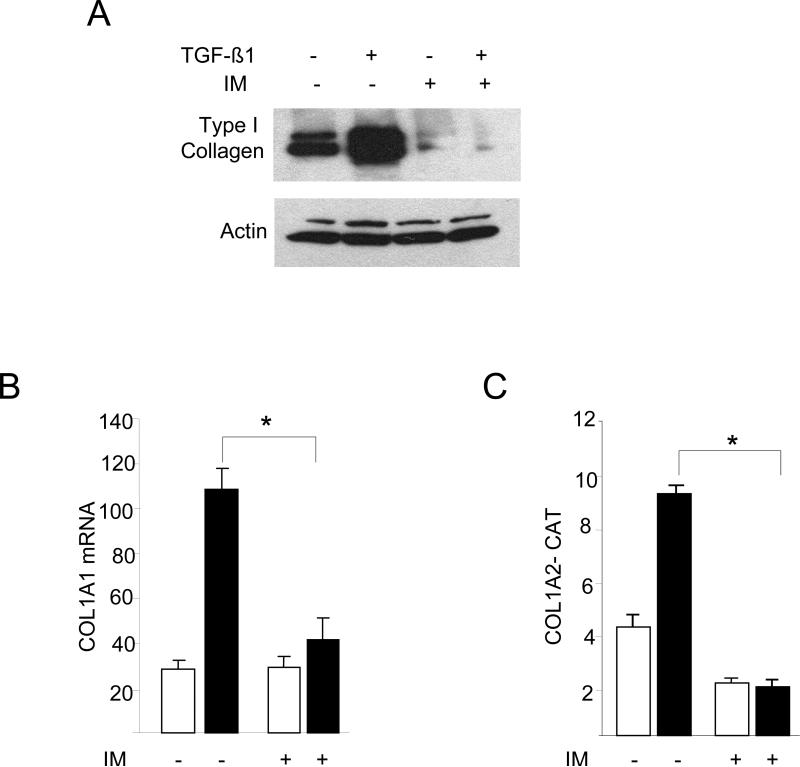
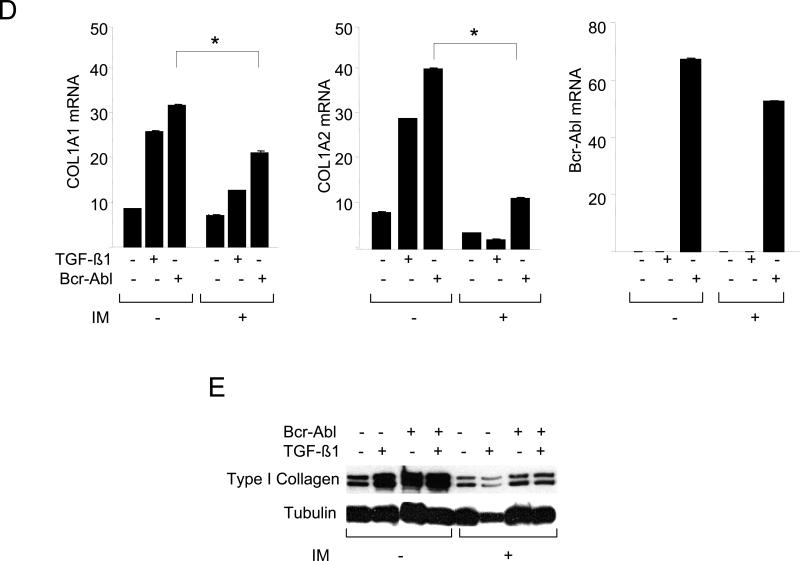
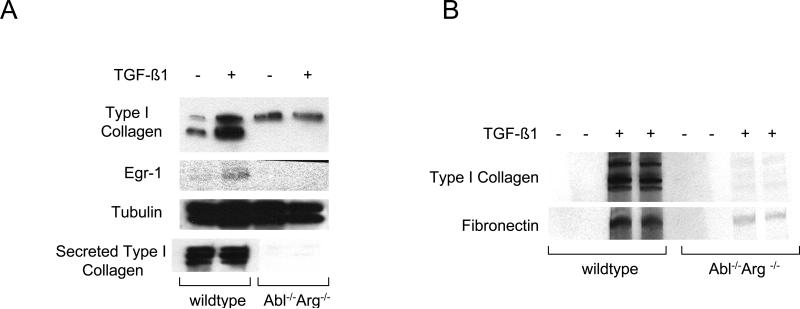
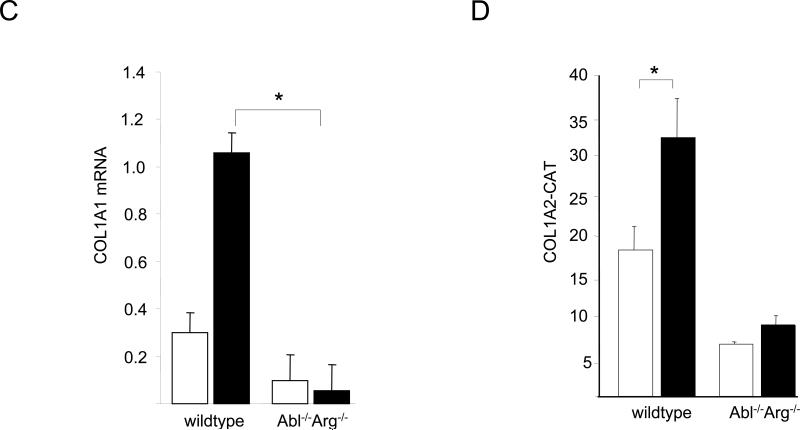
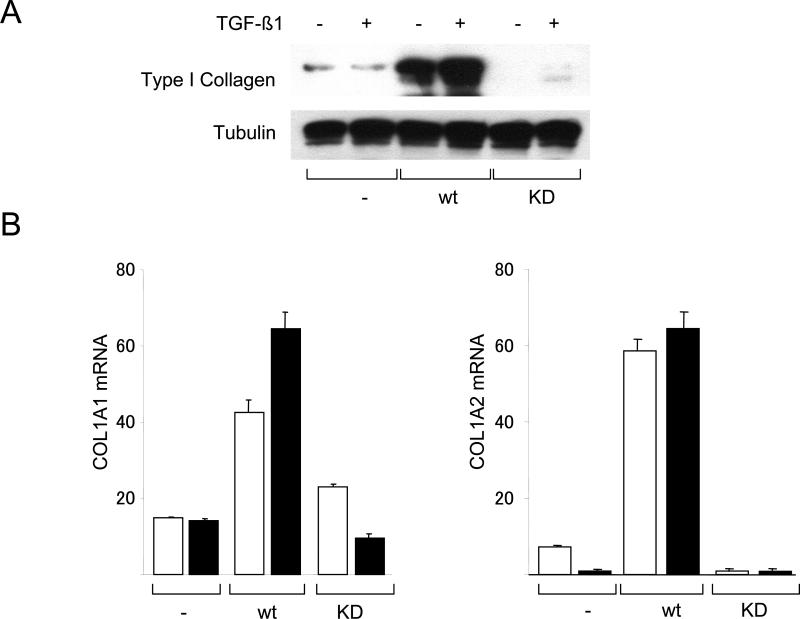
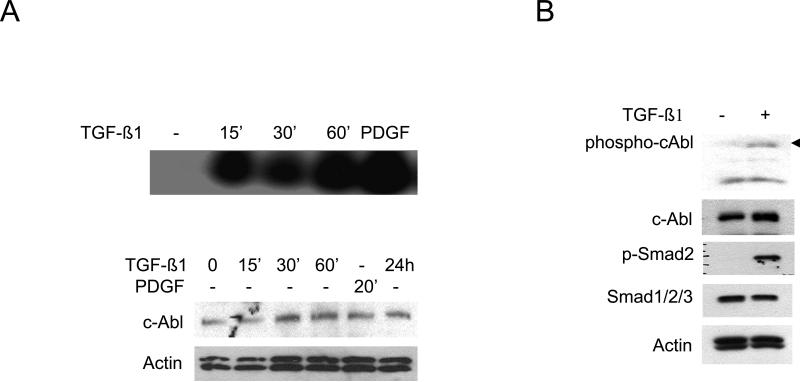
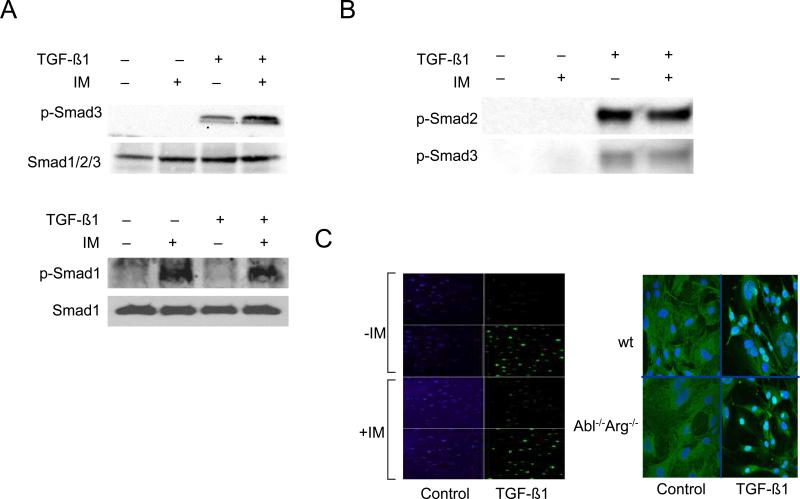
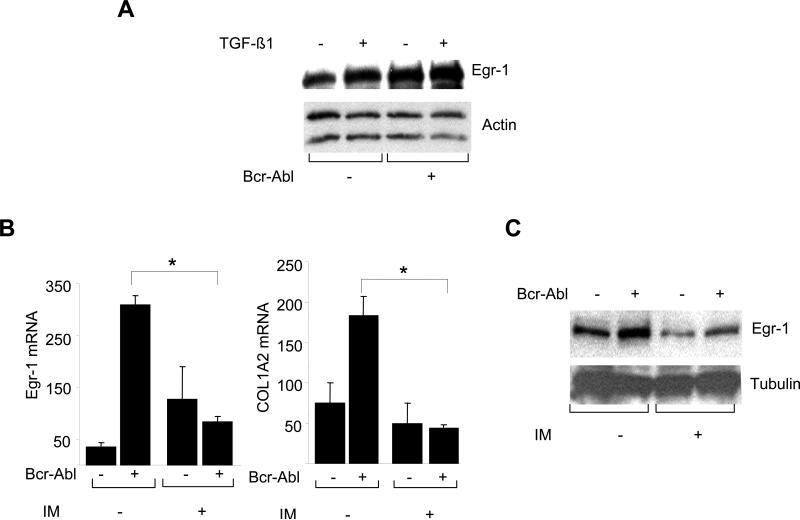
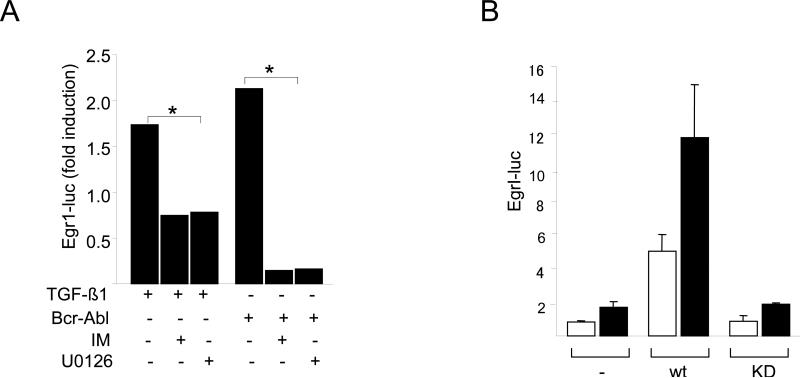
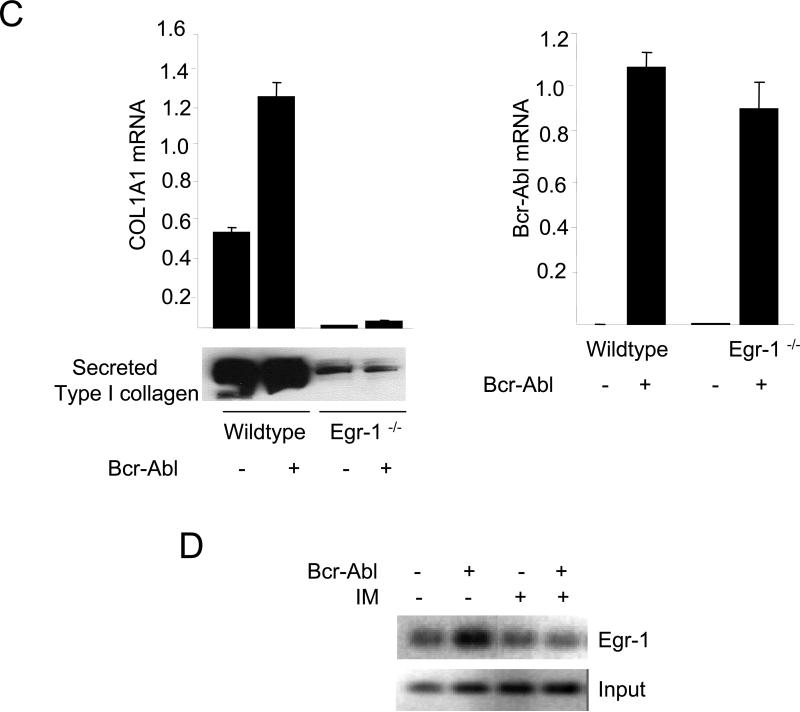
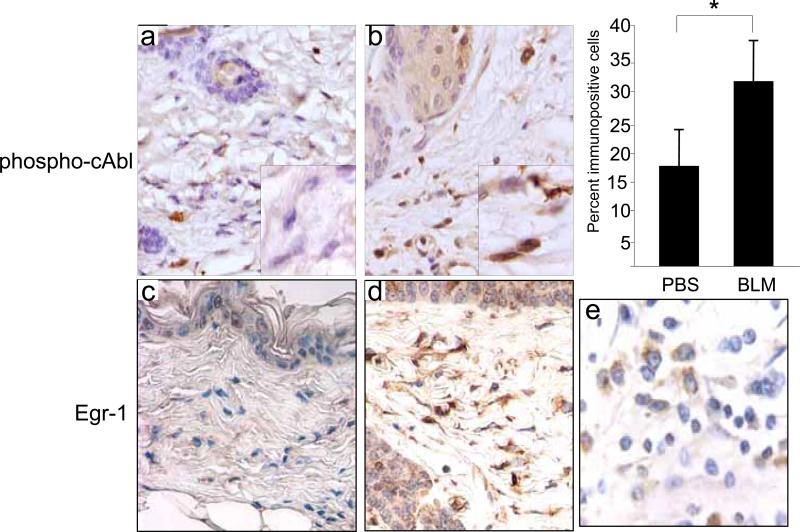
The nonreceptor protein tyrosine kinase c-Abl regulates cell proliferation and survival. Recent studies provide evidence that implicate c-Abl as a mediator for fibrotic responses induced by transforming growth factor-beta (TGF-beta), but the precise mechanisms underlying this novel oncogene function are unknown. Here, we report that when expressed in normal fibroblasts, a constitutively active mutant of Abl that causes chronic myelogenous leukemia (CML) stimulated the expression and transcriptional activity of the early growth response factor 1 (Egr-1). Mouse embryonic fibroblasts (MEFs), lacking c-Abl, were resistant to TGF-beta stimulation. Responsiveness of these MEFs to TGF-beta could be rescued by wild-type c-Abl, but not by a kinase-deficient mutant form of c-Abl. Furthermore, Abl kinase activity was necessary for the induction of Egr-1 by TGF-beta in normal fibroblasts, and Egr-1 was required for stimulation of collagen by Bcr-Abl. Lesional skin fibroblasts in mice with bleomycin-induced fibrosis of skin displayed evidence of c-Abl activation in situ, and elevated phospho-c-Abl correlated with increased local expression of Egr-1. Collectively, these results position Egr-1 downstream of c-Abl in the fibrotic response, delineate a novel Egr-1-dependent intracellular signaling mechanism that underlies the involvement of c-Abl in certain TGF-beta responses, and identify Egr-1 as a target of inhibition by imatinib. Furthermore, the findings show in situ activation of c-Abl paralleling the upregulated tissue expression of Egr-1 that accompanies fibrosis. Pharmacological targeting of c-Abl and its downstream effector pathways may, therefore, represent a novel therapeutic approach to blocking TGF-beta-dependent fibrotic processes.
Figures
References
-
- Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–45. - PubMed
-
- Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. - PubMed
-
- Moustakas A, Heldin CH. Non-Javelaud D, Mauviel. A Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005 Aug 29;24(37):5742–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
