

The molecular and cellular biology of enhanced cognition
- PMID: 19153576
- PMCID: PMC2664745
- DOI: 10.1038/nrn2572
The molecular and cellular biology of enhanced cognition
Abstract
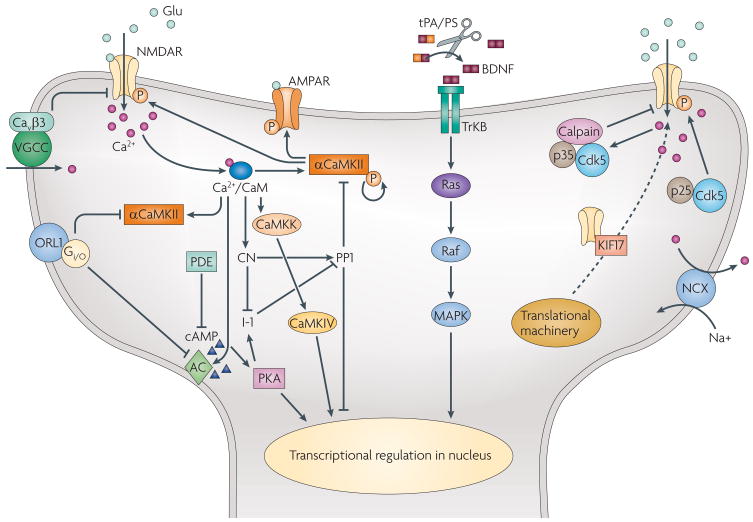
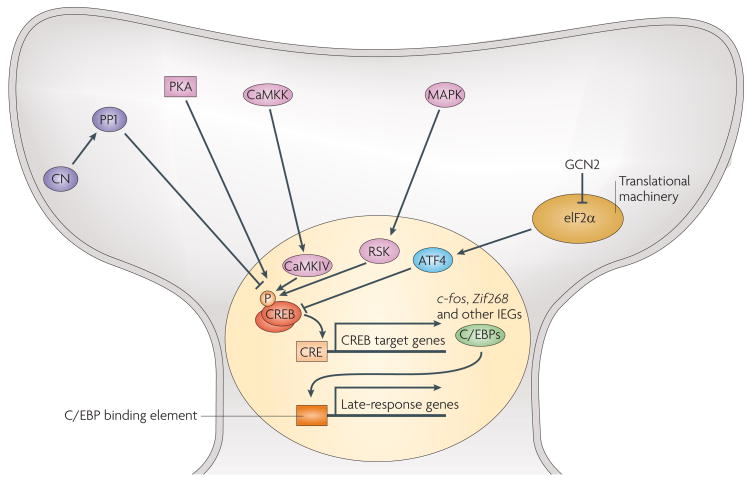
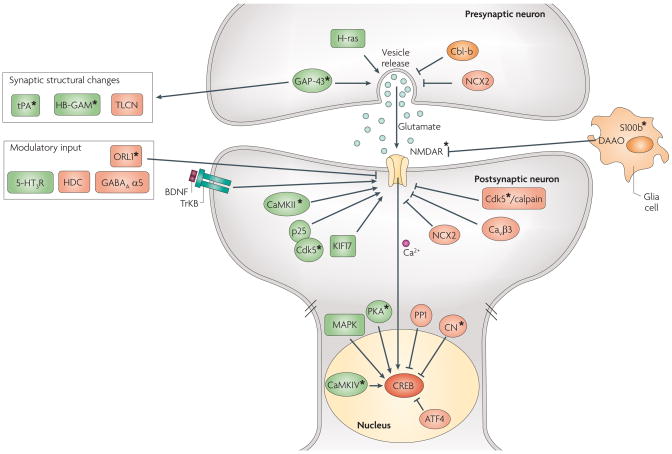
Most molecular and cellular studies of cognitive function have focused on either normal or pathological states, but recent research with transgenic mice has started to address the mechanisms of enhanced cognition. These results point to key synaptic and nuclear signalling events that can be manipulated to facilitate the induction or increase the stability of synaptic plasticity, and therefore enhance the acquisition or retention of information. Here, we review these surprising findings and explore their implications to both mechanisms of learning and memory and to ongoing efforts to develop treatments for cognitive disorders. These findings represent the beginning of a fundamental new approach in the study of enhanced cognition.
Figures
References
-
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. - PubMed
-
- Harwood AJ, Agam G. Search for a common mechanism of mood stabilizers. Biochem Pharmacol. 2003;66:179–189. - PubMed
-
- Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Suppl 2):183–194. - PubMed
-
- Costa RM, Silva AJ. Molecular and cellular mechanisms underlying the cognitive deficits associated with neurofibromatosis 1. J Child Neurol. 2002;17:622–626. discussion 627–629, 646–651. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
