

On the molecular etiology of Cornelia de Lange syndrome
- PMID: 19154515
- PMCID: PMC2733214
- DOI: 10.1111/j.1749-6632.2008.03450.x
On the molecular etiology of Cornelia de Lange syndrome
Abstract
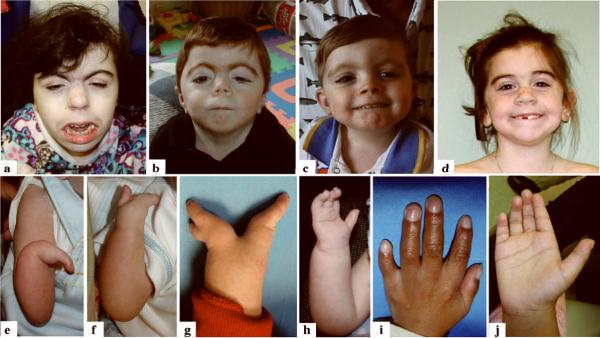
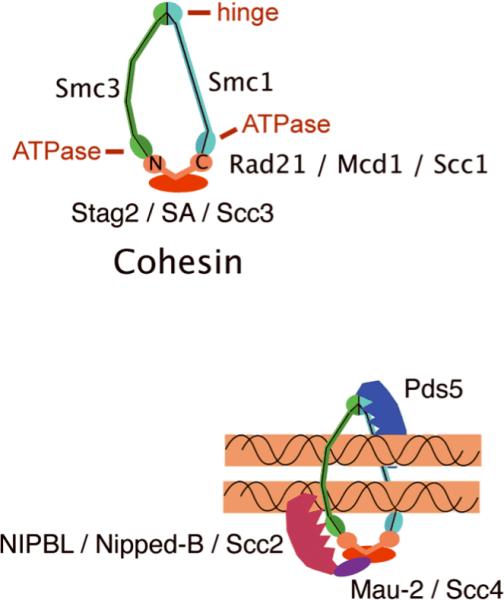
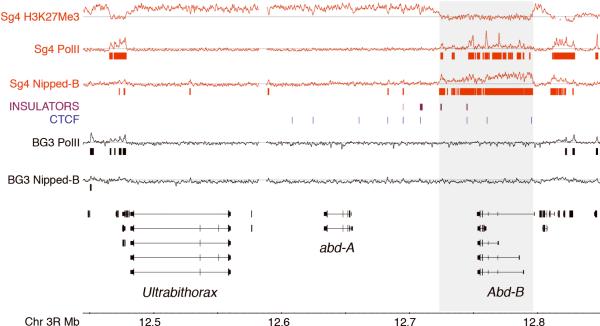
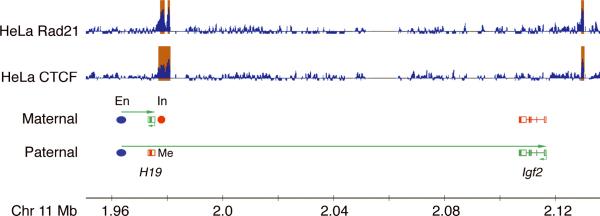
Cornelia de Lange syndrome (CdLS) is genetically heterogeneous and is usually sporadic, occurring approximately once per 10,000 births. CdLS individuals display diverse and variable deficits in growth, mental development, limbs, and organs. In the past few years it has been shown that CdLS is caused by gene mutations affecting proteins involved in sister chromatid cohesion. Studies in model organisms, and more recently in human cells, have revealed, somewhat unexpectedly, that the developmental deficits in CdLS likely arise from changes in gene expression. The mechanisms by which cohesion factors regulate gene expression remain to be elucidated, but current data suggest that they likely regulate transcription in multiple ways.
Figures
References
-
- Vrolik W. Tabulae ad illustrandam embryogenesin hominis et mammalium tam naturalem quam abnormem. Londonck; Amsterdam: 1849.
-
- Brachmann W. Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnormitaten (zwerghaftogkeit, halsrippen, behaarung) Jarb Kinder Phys Erzie. 1916;84:225–35.
-
- de Lange C. Sur un type nouveau de dégénération (typus Amstelodamensis) Arch Méd Enfants. 1933;36:713–19.
-
- Ireland M, Donnai D, Burn J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am J Med Genet. 1993;47:959–63. - PubMed
-
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: a clinical review of 310 individuals. Am J Med Genet. 1993;47:940–46. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
