

Molecular Insight into the Synergism between the Minor Allele of Human Liver Peroxisomal Alanine:Glyoxylate Aminotransferase and the F152I Mutation
- PMID: 19155213
- PMCID: PMC2659193
- DOI: 10.1074/jbc.M808965200
Molecular Insight into the Synergism between the Minor Allele of Human Liver Peroxisomal Alanine:Glyoxylate Aminotransferase and the F152I Mutation
Abstract
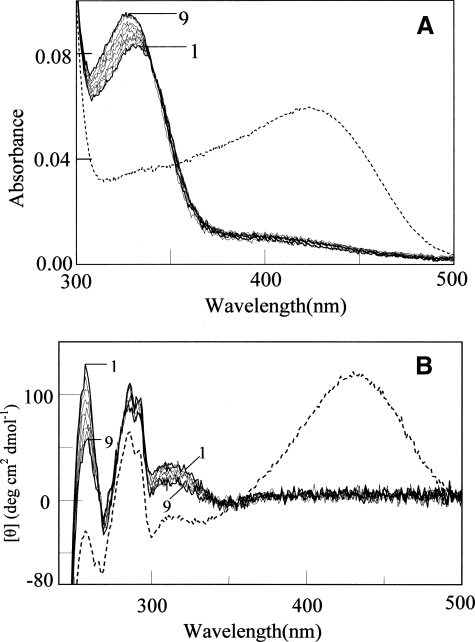
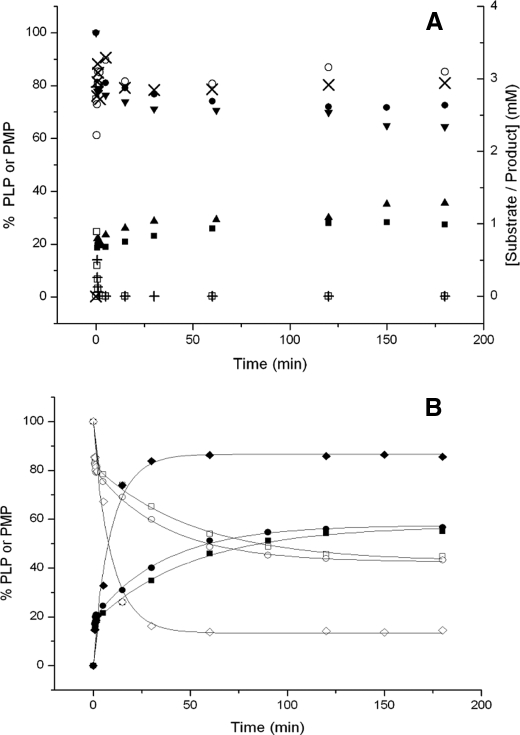
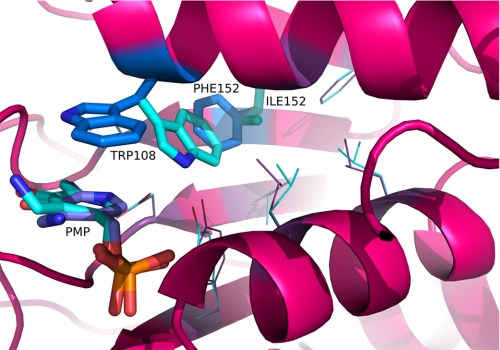
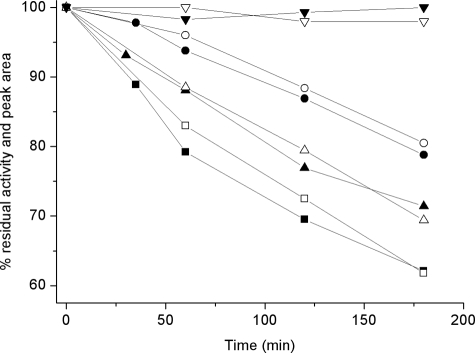
Human liver peroxisomal alanine:glyoxylate aminotransferase (AGT) is a pyridoxal 5'-phosphate (PLP)-dependent enzyme that converts glyoxylate into glycine. AGT deficiency causes primary hyperoxaluria type 1 (PH1), a rare autosomal recessive disorder, due to a marked increase in hepatic oxalate production. Normal human AGT exists as two polymorphic variants: the major (AGT-Ma) and the minor (AGT-Mi) allele. AGT-Mi causes the PH1 disease only when combined with some mutations. In this study, the molecular basis of the synergism between AGT-Mi and F152I mutation has been investigated through a detailed biochemical characterization of AGT-Mi and the Phe(152) variants combined either with the major (F152I-Ma, F152A-Ma) or the minor allele (F152I-Mi). Although these species show spectral features, kinetic parameters, and PLP binding affinity similar to those of AGT-Ma, the Phe(152) variants exhibit the following differences with respect to AGT-Ma and AGT-Mi: (i) pyridoxamine 5'-phosphate (PMP) is released during the overall transamination leading to the conversion into apoenzymes, and (ii) the PMP binding affinity is at least 200-1400-fold lower. Thus, Phe(152) is not an essential residue for transaminase activity, but plays a role in selectively stabilizing the AGT-PMP complex, by a proper orientation of Trp(108), as suggested by bioinformatic analysis. These data, together with the finding that apoF152I-Mi is the only species that at physiological temperature undergoes a time-dependent inactivation and concomitant aggregation, shed light on the molecular defects resulting from the association of the F152I mutation with AGT-Mi, and allow to speculate on the responsiveness to pyridoxine therapy of PH1 patients carrying this mutation.
Figures

Similar articles
-
Human liver peroxisomal alanine:glyoxylate aminotransferase: characterization of the two allelic forms and their pathogenic variants.Biochim Biophys Acta. 2011 Nov;1814(11):1577-84. doi: 10.1016/j.bbapap.2010.12.005. Epub 2010 Dec 20. Biochim Biophys Acta. 2011. PMID: 21176891 Review.
-
Misfolding caused by the pathogenic mutation G47R on the minor allele of alanine:glyoxylate aminotransferase and chaperoning activity of pyridoxine.Biochim Biophys Acta. 2015 Oct;1854(10 Pt A):1280-9. doi: 10.1016/j.bbapap.2015.07.002. Epub 2015 Jul 3. Biochim Biophys Acta. 2015. PMID: 26149463
-
Human liver peroxisomal alanine:glyoxylate aminotransferase: Different stability under chemical stress of the major allele, the minor allele, and its pathogenic G170R variant.Biochimie. 2010 Dec;92(12):1801-11. doi: 10.1016/j.biochi.2010.08.005. Epub 2010 Aug 14. Biochimie. 2010. PMID: 20713123
-
Human wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: functional properties and physiological implications.Biochem J. 2007 Nov 15;408(1):39-50. doi: 10.1042/BJ20070637. Biochem J. 2007. PMID: 17696873 Free PMC article.
-
Liver peroxisomal alanine:glyoxylate aminotransferase and the effects of mutations associated with Primary Hyperoxaluria Type I: An overview.Biochim Biophys Acta. 2015 Sep;1854(9):1212-9. doi: 10.1016/j.bbapap.2014.12.029. Epub 2015 Jan 22. Biochim Biophys Acta. 2015. PMID: 25620715 Review.
Cited by
-
CLYBL is a polymorphic human enzyme with malate synthase and β-methylmalate synthase activity.Hum Mol Genet. 2014 May 1;23(9):2313-23. doi: 10.1093/hmg/ddt624. Epub 2013 Dec 11. Hum Mol Genet. 2014. PMID: 24334609 Free PMC article.
-
Identification of Human Alanine-Glyoxylate Aminotransferase Ligands as Pharmacological Chaperones for Variants Associated with Primary Hyperoxaluria Type 1.J Med Chem. 2022 Jul 28;65(14):9718-9734. doi: 10.1021/acs.jmedchem.2c00142. Epub 2022 Jul 13. J Med Chem. 2022. PMID: 35830169 Free PMC article.
-
Molecular therapy of primary hyperoxaluria.J Inherit Metab Dis. 2017 Jul;40(4):481-489. doi: 10.1007/s10545-017-0045-3. Epub 2017 Apr 19. J Inherit Metab Dis. 2017. PMID: 28425073 Review.
-
Crystal structure of the S187F variant of human liver alanine: glyoxylate [corrected] aminotransferase associated with primary hyperoxaluria type I and its functional implications.Proteins. 2013 Aug;81(8):1457-65. doi: 10.1002/prot.24300. Epub 2013 Jun 1. Proteins. 2013. PMID: 23589421 Free PMC article.
-
A Minor Haplotype Variant Determines the Pathogenicity of the p.Ile279Thr Substitution in the Primary Hyperoxaluria Type 1 Gene, AGXT.J Inherit Metab Dis. 2025 Jul;48(4):e70052. doi: 10.1002/jimd.70052. J Inherit Metab Dis. 2025. PMID: 40495747 Free PMC article.
References
-
- Danpure, C. J., Fryer, P., Griffiths, S., Guttridge, K. M., Jennings, P. R., Allsop, J., Moser, A. B., Naidu, S., Moser, H. W., MacCollin, M., and Devino, D. C. (1994) J. Inherit Metab. Dis. 17 27–40 - PubMed
-
- Lumb, M. J., and Danpure, C. J. (2000) J. Biol. Chem. 275 36415–36422 - PubMed
-
- Zhang, X., Roe, S. M., Hou, Y., Bartlam, M., Rao, Z., Pearl, L. H., and Danpure, C. J. (2003) J. Mol. Biol. 331 643–652 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
