

Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond
- PMID: 19157957
- PMCID: PMC2723731
- DOI: 10.1016/j.pbi.2008.11.004
Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond
Abstract
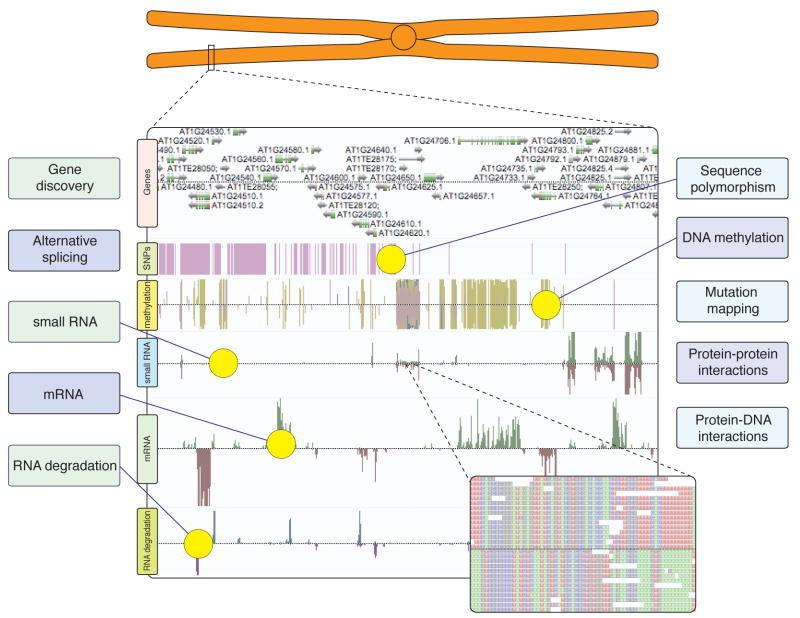
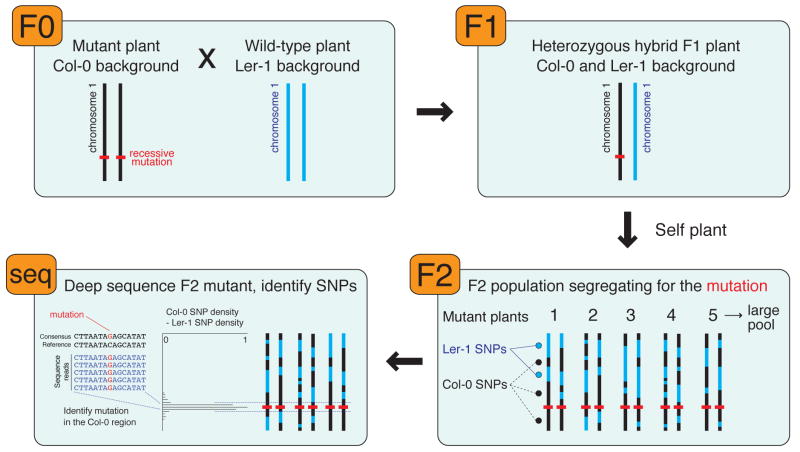
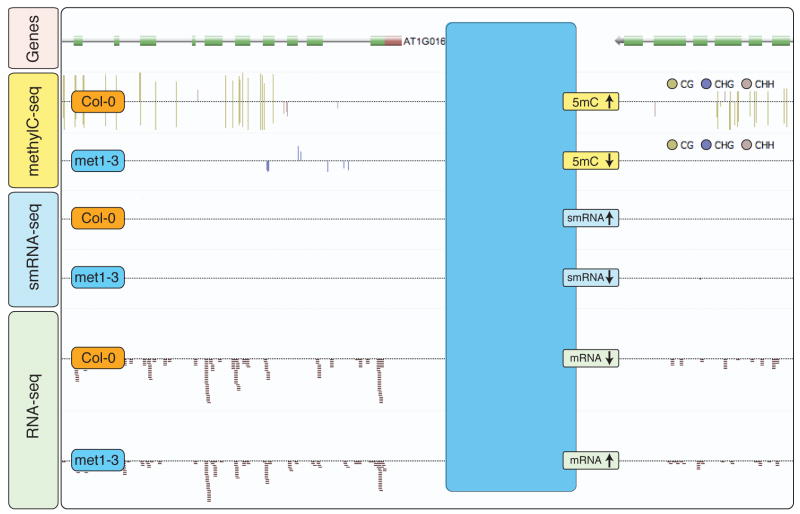
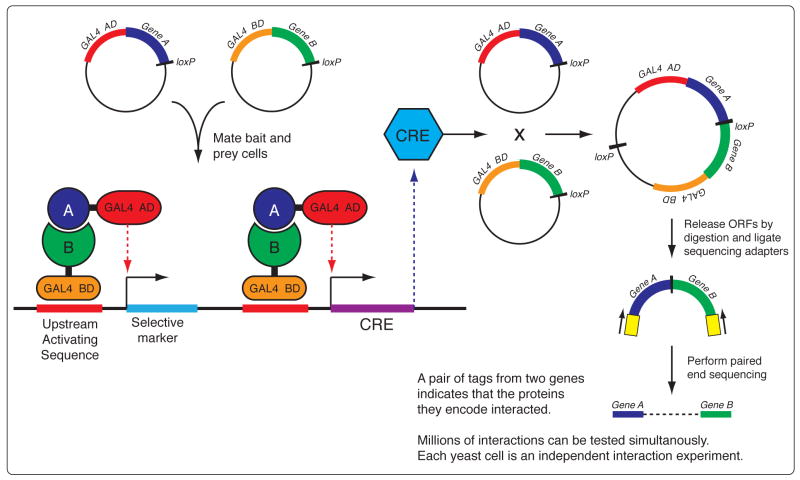
The sudden availability of DNA sequencing technologies that rapidly produce vast amounts of sequence information has triggered a paradigm shift in genomics, enabling massively parallel surveying of complex nucleic acid populations. The diversity of applications to which these technologies have already been applied demonstrates the immense range of cellular processes and properties that can now be studied at the single-base resolution. These include genome resequencing and polymorphism discovery, mutation mapping, DNA methylation, histone modifications, transcriptome sequencing, gene discovery, alternative splicing identification, small RNA profiling, DNA-protein, and possibly even protein-protein interactions. Thus, these deep sequencing technologies offer plant biologists unprecedented opportunities to increase the understanding of the functions and dynamics of plant cells and populations.
Conflict of interest statement
The authors declare that there are no conflicts of interest related to this publication.
Figures
References
-
- Mardis ER. Next-generation DNA sequencing methods. Annual review of genomics and human genetics. 2008;9:387–402. - PubMed
-
- Shendure JHJi. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. - PubMed
-
- Clark RM, Schweikert G, Toomajian C, Ossowski S, Zeller G, Shinn P, Warthmann N, Hu TT, Fu G, Hinds DA, et al. Common Sequence Polymorphisms Shaping Genetic Diversity in Arabidopsis thaliana. Science. 2007;317:338–342. - PubMed
-
- Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. - PubMed
-
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science. 2001;294:1719–1723. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
