

High prevalence of four long QT syndrome founder mutations in the Finnish population
- PMID: 19160088
- PMCID: PMC2704397
- DOI: 10.1080/07853890802668530
High prevalence of four long QT syndrome founder mutations in the Finnish population
Abstract
Aims: Long QT syndrome (LQTS) is an inherited arrhythmia disorder with an estimated prevalence of 0.01%-0.05%. In Finland, four founder mutations constitute up to 70% of the known genetic spectrum of LQTS. In the present survey, we sought to estimate the actual prevalence of the founder mutations and to determine their effect sizes in the general Finnish population.
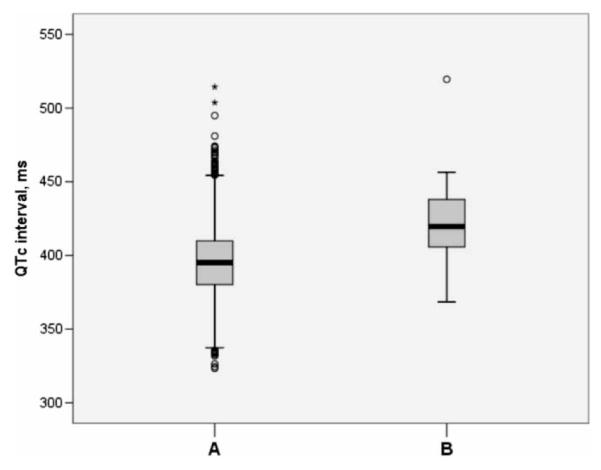
Methods and results: We genotyped 6334 subjects aged > or =30 years from a population cohort (Health 2000 study) for the four Finnish founder mutations using Sequenom MALDI-TOF mass spectrometry. The electrocardiogram (ECG) parameters were measured from digital 12-lead ECGs, and QT intervals were adjusted for age, sex, and heart rate using linear regression. A total of 27 individuals carried one of the founder mutations resulting in their collective prevalence estimate of 0.4% (95% CI 0.3%-0.6%). The KCNQ1 G589D mutation (n=8) was associated with a 50 ms (SE 7.0) prolongation of the adjusted QT interval (P=9.0x10(-13)). The KCNH2 R176W variant (n=16) resulted in a 22 ms (SE 4.7) longer adjusted QT interval (P=2.1x10(-6)).
Conclusion: In Finland 1 individual out of 250 carries a LQTS founder mutation, which is the highest documented prevalence of LQTS mutations that lead to a marked QT prolongation.
Figures
References
-
- Viskin S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–33. - PubMed
-
- Priori SG, Napolitano C, Schwartz PJ. Genetics of cardiac arrhythmias. In: Libby P, Bonow RO, Mann DL, Zipes DP, editors. Braunwald’s Heart Disease. 8th ed Saunders Elsevier; Philadelphia, PA: 2008.
-
- Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47. - PubMed
-
- Stramba-Badiale M, Crotti L, Goulene K, Pedrazzini M, Mannarino S, Salice P, et al. Electrocardiographic and genetic screening for long QT syndrome: results from a prospective study on 44,596 neonates. Circulation. 2007;116 Supplement:II_377.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources