

Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD)
- PMID: 19161632
- PMCID: PMC2635369
- DOI: 10.1186/1471-230X-9-8
Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD)
Abstract
Background: Congenital lactase deficiency (CLD) is a severe gastrointestinal disorder of newborns. The diagnosis is challenging and based on clinical symptoms and low lactase activity in intestinal biopsy specimens. The disease is enriched in Finland but is also present in other parts of the world. Mutations encoding the lactase (LCT) gene have recently been shown to underlie CLD. The purpose of this study was to identify new mutations underlying CLD in patients with different ethnic origins, and to increase awareness of this disease so that the patients could be sought out and treated correctly.
Methods: Disaccharidase activities in intestinal biopsy specimens were assayed and the coding region of LCT was sequenced from five patients from Europe with clinical features compatible with CLD. In the analysis and prediction of mutations the following programs: ClustalW, Blosum62, PolyPhen, SIFT and Panther PSEC were used.
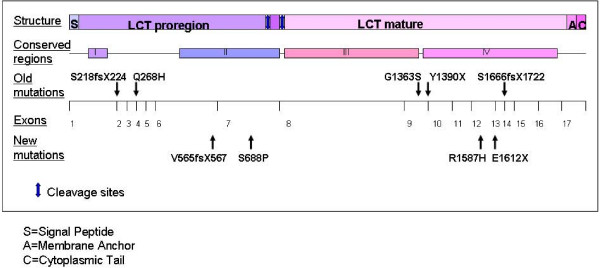
Results: Four novel mutations in the LCT gene were identified. A single nucleotide substitution leading to an amino acid change S688P in exon 7 and E1612X in exon 12 were present in a patient of Italian origin. Five base deletion V565fsX567 leading to a stop codon in exon 6 was found in one and a substitution R1587H in exon 12 from another Finnish patient. Both Finnish patients were heterozygous for the Finnish founder mutation Y1390X. The previously reported mutation G1363S was found in a homozygous state in two siblings of Turkish origin.
Conclusion: This is the first report of CLD mutations in patients living outside Finland. It seems that disease is more common than previously thought. All mutations in the LCT gene lead to a similar phenotype despite the location and/or type of mutation.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
