

Response gene to complement 32 is required for C5b-9 induced cell cycle activation in endothelial cells
- PMID: 19162005
- PMCID: PMC2699899
- DOI: 10.1016/j.yexmp.2008.12.005
Response gene to complement 32 is required for C5b-9 induced cell cycle activation in endothelial cells
Abstract
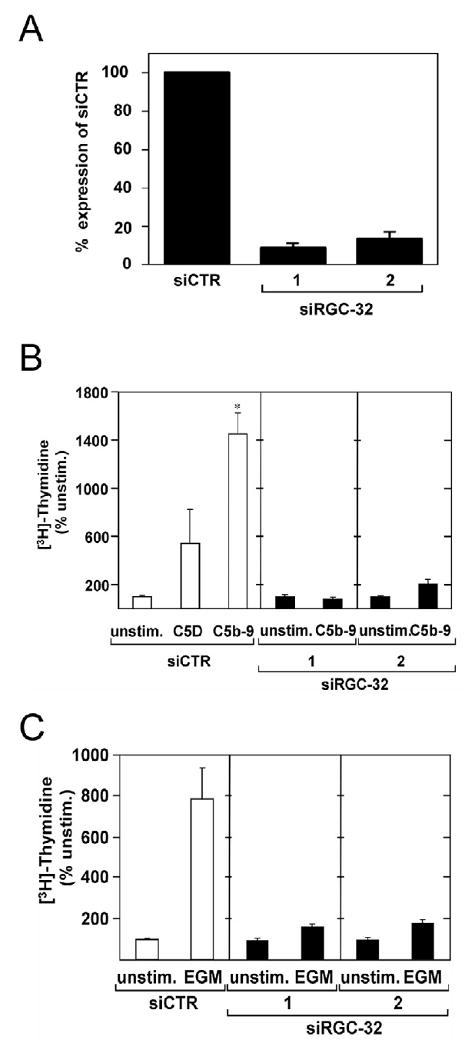
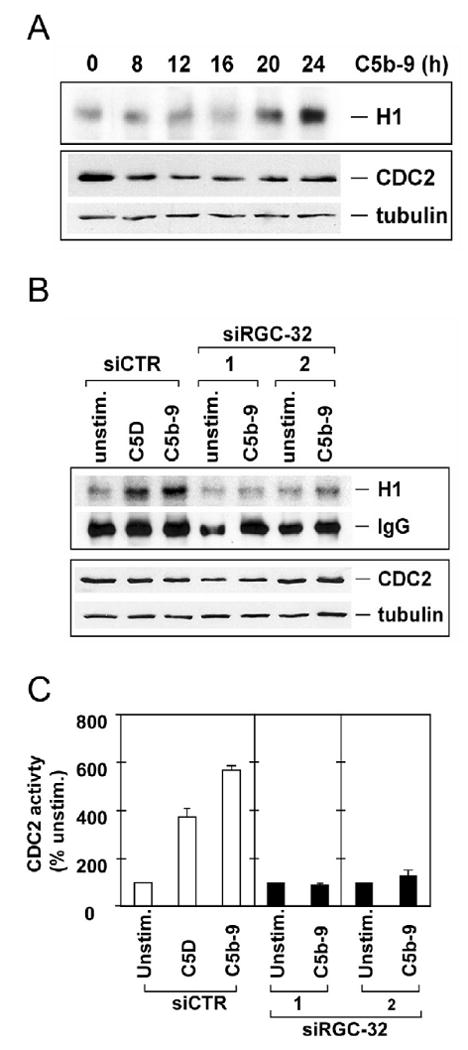
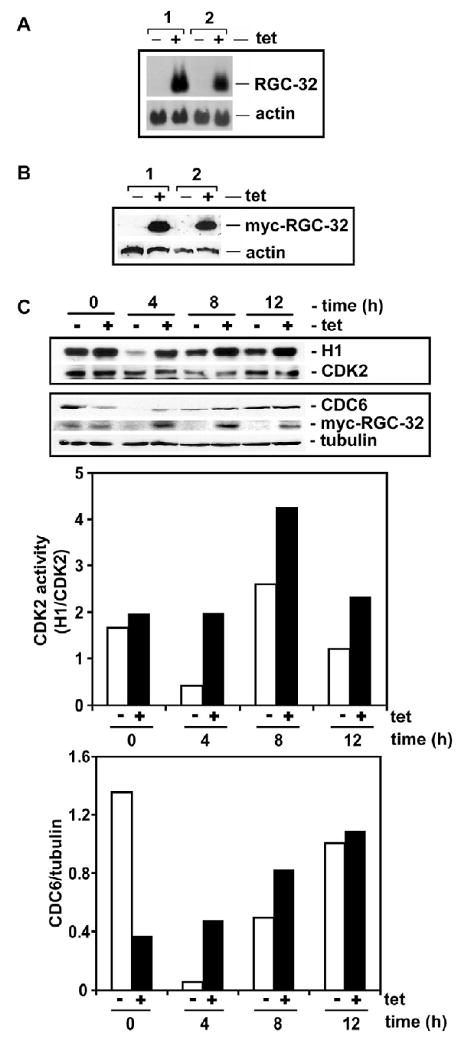
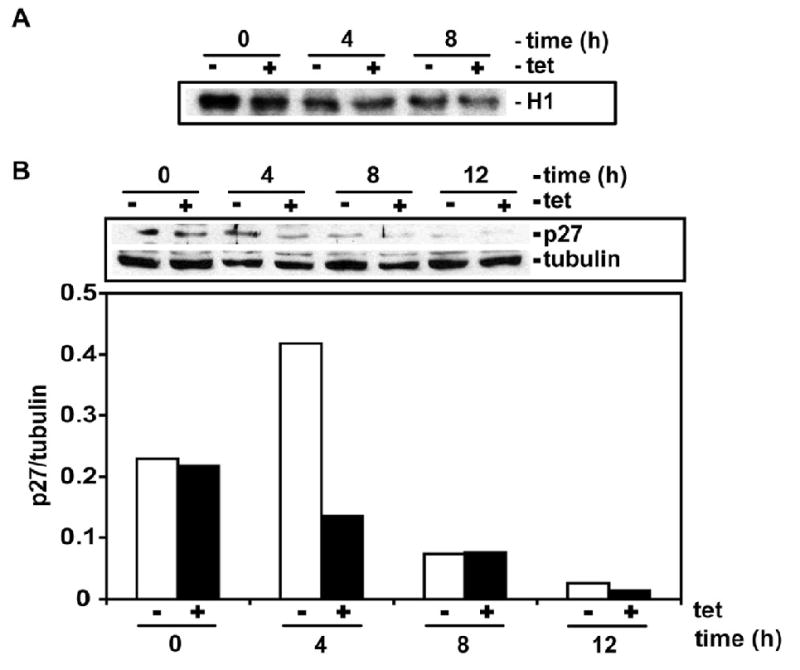
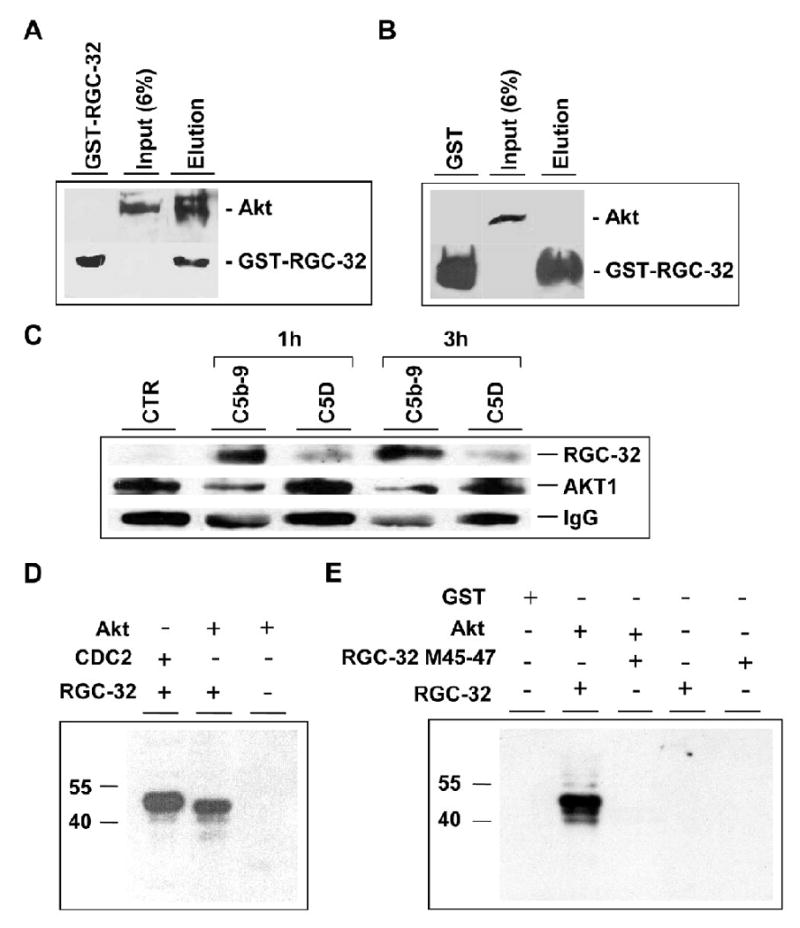
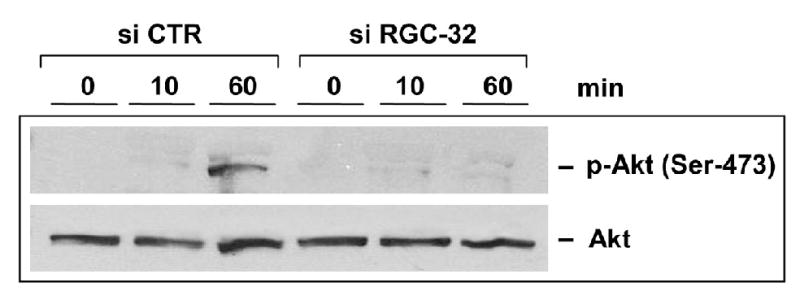
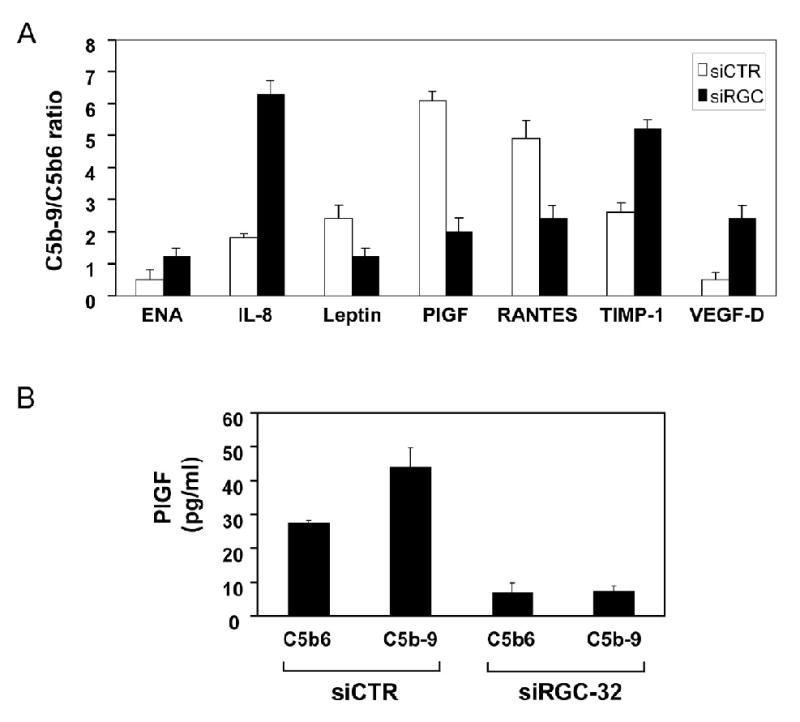
Proliferation of vascular endothelial cells (EC) and smooth muscle cells (SMC) is a critical event in angiogenesis and atherosclerosis. We previously showed that the C5b-9 assembly during complement activation induces cell cycle in human aortic EC (AEC) and SMC. C5b-9 can induce the expression of Response Gene to Complement (RGC)-32 and over expression of this gene leads to cell cycle activation. Therefore, the present study was carried out to test the requirement of endogenous RGC-32 for the cell cycle activation induced by C5b-9 by knocking-down its expression using siRNA. We identified two RGC-32 siRNAs that can markedly reduce the expression of RGC-32 mRNA in AEC. RGC-32 silencing in these cells abolished DNA synthesis induced by C5b-9 and serum growth factors, indicating the requirement of RGC-32 activity for S-phase entry. RGC-32 siRNA knockdown also significantly reduced the C5b-9 induced CDC2 activation and Akt phosphorylation. CDC2 does not play a role in G1/S transition in HeLa cells stably overexpressing RGC-32. RGC-32 was found to physically associate with Akt and was phosphorylated by Akt in vitro. Mutation of RGC-32 protein at Ser 45 and Ser 47 prevented Akt mediated phosphorylation. In addition, RGC-32 was found to regulate the release of growth factors from AEC. All these data together suggest that cell cycle induction by C5b-9 in AEC is RGC-32 dependent and this is in part through regulation of Akt and growth factor release.
Figures
References
-
- Badea T, Niculescu F, Soane L, Fosbrink M, Sorana H, Rus V, Shin ML, Rus H. RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase. J Biol Chem. 2002;277:502–8. - PubMed
-
- Badea TC, Niculescu FI, Soane L, Shin ML, Rus H. Molecular cloning and characterization of RGC-32, a novel gene induced by complement activation in oligodendrocytes. J Biol Chem. 1998;273:26977–81. - PubMed
-
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. - PubMed
-
- Carney DF, Koski CL, Shin ML. Elimination of terminal complement intermediates from the plasma membrane of nucleated cells: the rate of disappearance differs for cells carrying C5b-7 or C5b-8 or a mixture of C5b-8 with a limited number of C5b-9. J Immunol. 1985;134:1804–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
