

Computational evaluation of protein-small molecule binding
- PMID: 19162472
- PMCID: PMC2671234
- DOI: 10.1016/j.sbi.2008.11.009
Computational evaluation of protein-small molecule binding
Abstract
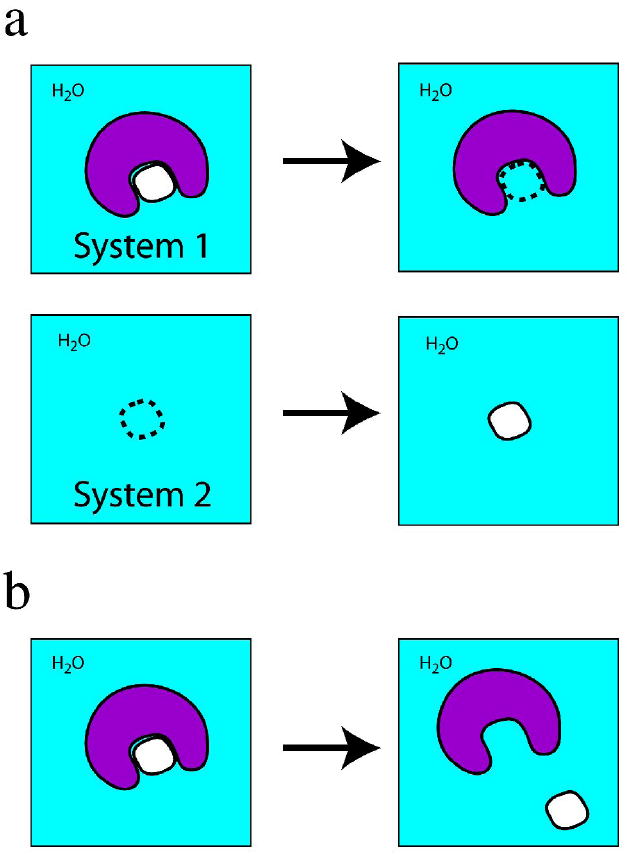
Determining protein-small molecule binding affinity is a key component of present-day rational drug discovery. To circumvent the time, labor, and materials costs associated with experimental protein-small molecule binding assays, a variety of structure-based computational methods have been developed for determining protein-small molecule binding affinities. These methods can be placed in one of two classes: accurate but slow (Class 1), and fast but approximate (Class 2). Class 1 methods, which explicitly take into account protein flexibility and include an atomic-level description of solvation, are capable of quantitatively reproducing experimental protein-small molecule absolute binding free energies. However, Class 1 computational requirements make screening thousands to millions of small molecules against a protein, as required for rational drug design, infeasible for the foreseeable future. Class 2 methods, on the contrary, are sufficiently fast to perform such inhibitor screening, yet they suffer from limited descriptions of protein flexibility and solvation, which in turn limit their ability to select and rank-order small molecules by computed binding affinities. This review presents an overview of Class 1 and Class 2 methods, and avenues of research in Class 2 methods aimed at bringing them closer to Class 1 accuracy.
Figures
References
-
- Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nature Structural Biology. 2002;9:646–652. - PubMed
-
- MacKerell AD., Jr Empirical force fields for biological macromolecules: overview and issues. Journal Of Computational Chemistry. 2004;25:1584–1604. - PubMed
-
- Guvench O, MacKerell AD. Comparison of protein force fields for molecular dynamics simulations. In: Kukol A, editor. Molecular Modeling of Proteins. Humana Press, Inc.; 2008. pp. 63–88. Methods in Molecular Biology. - PubMed
-
- Tuckerman ME, Martyna GJ. Understanding modern molecular dynamics: Techniques and applications. Journal of Physical Chemistry B. 2000;104:159–178.
Publication types
MeSH terms
Substances
Grants and funding
- GM51501/GM/NIGMS NIH HHS/United States
- HL082670/HL/NHLBI NIH HHS/United States
- CA107331/CA/NCI NIH HHS/United States
- CA120215/CA/NCI NIH HHS/United States
- R01 CA120215/CA/NCI NIH HHS/United States
- R21 HL082670/HL/NHLBI NIH HHS/United States
- R01 GM072558/GM/NIGMS NIH HHS/United States
- R01 GM070855/GM/NIGMS NIH HHS/United States
- R01 CA107331/CA/NCI NIH HHS/United States
- F32CA1197712/CA/NCI NIH HHS/United States
- R01 GM051501/GM/NIGMS NIH HHS/United States
- R29 GM051501/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
