

Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice
- PMID: 19164484
- PMCID: PMC2670667
- DOI: 10.1152/ajpgi.90520.2008
Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice
Abstract
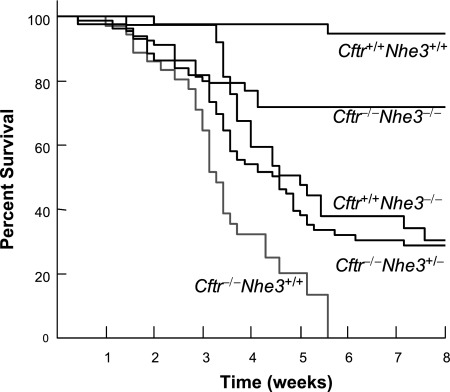
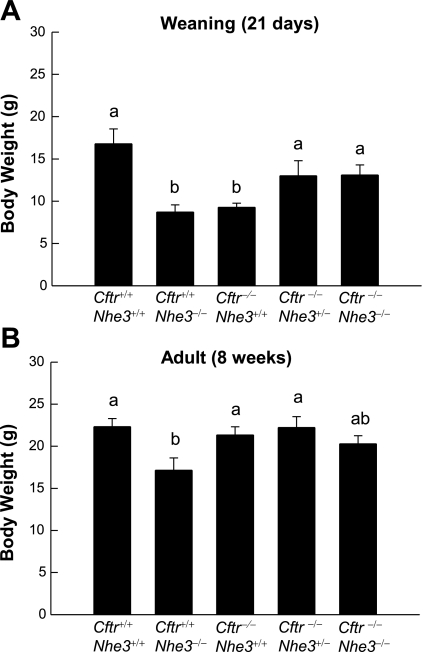
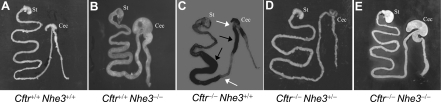
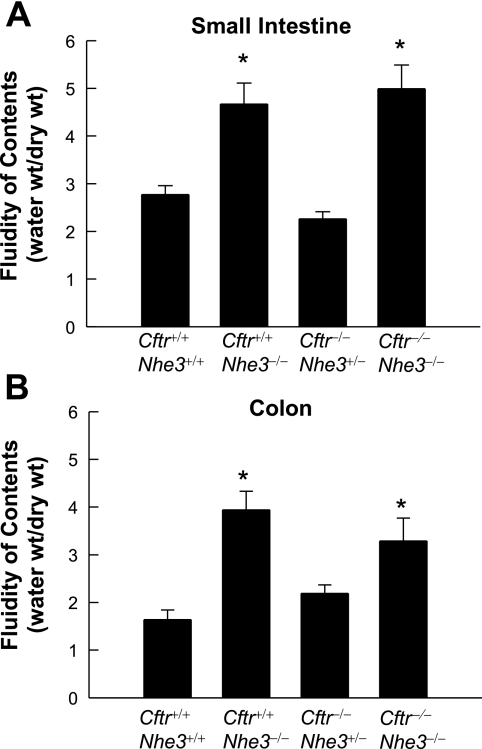
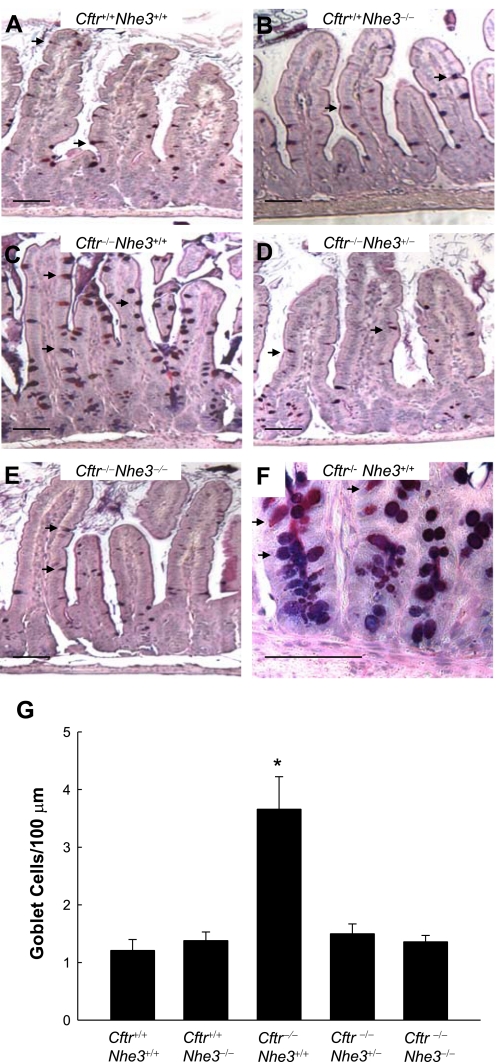
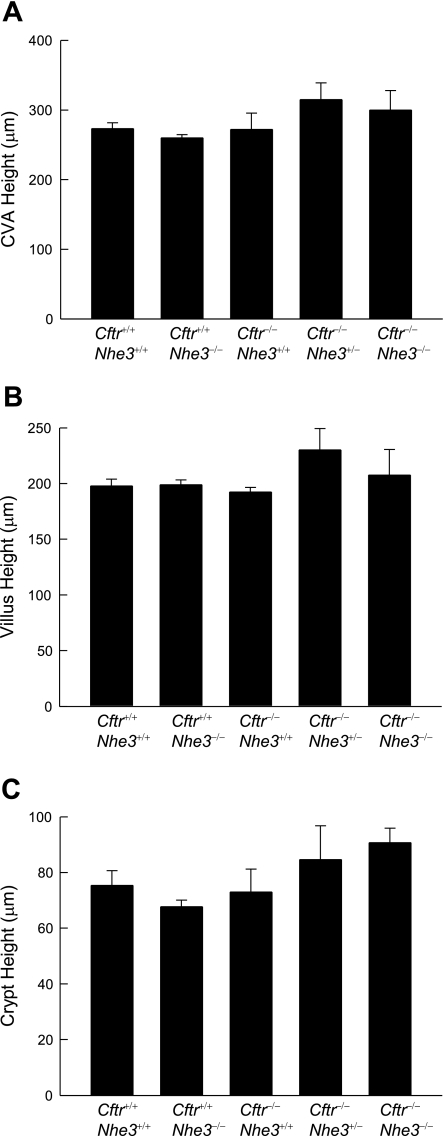
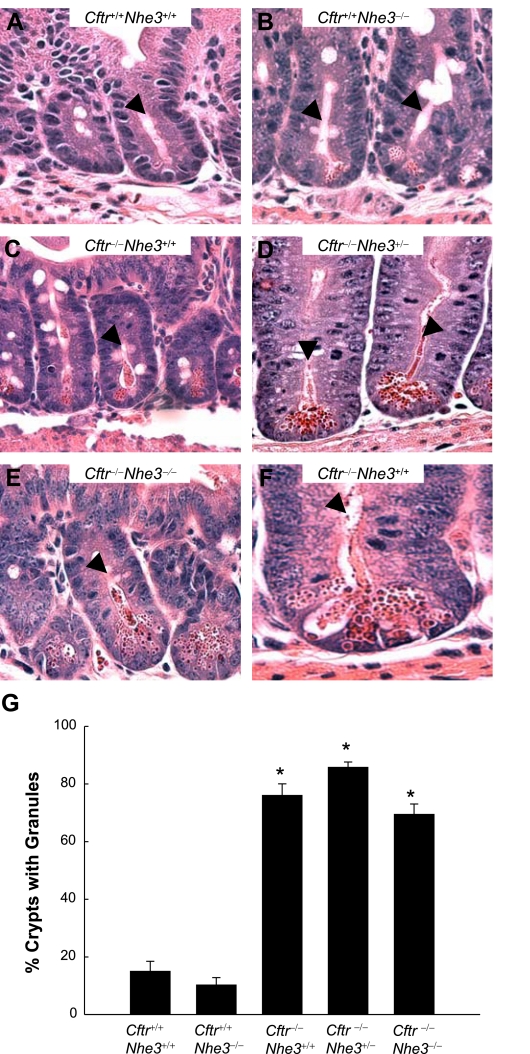
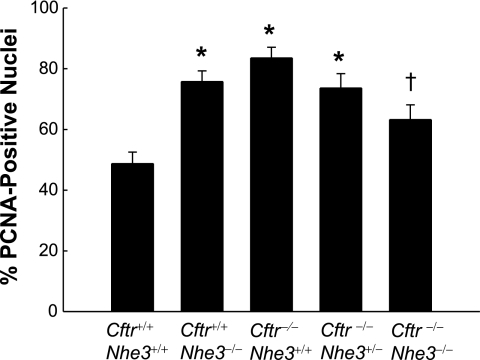
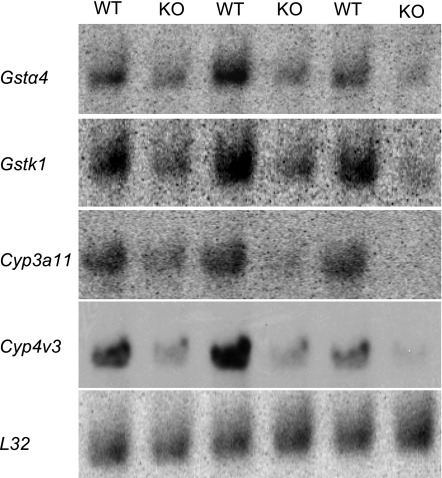
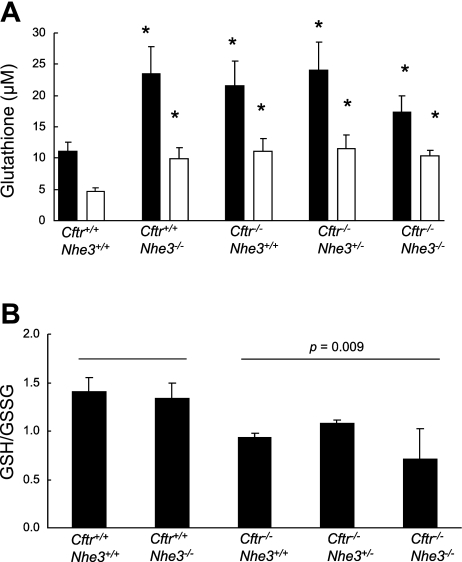
In cystic fibrosis, impaired secretion resulting from loss of activity of the cystic fibrosis transmembrane conductance regulator (CFTR) causes dehydration of intestinal contents and life-threatening obstructions. Conversely, impaired absorption resulting from loss of the NHE3 Na+/H+ exchanger causes increased fluidity of the intestinal contents and diarrhea. To test the hypothesis that reduced NHE3-mediated absorption could increase survival and prevent some of the intestinal pathologies of cystic fibrosis, Cftr/Nhe3 double heterozygous mice were mated and their offspring analyzed. Cftr-null mice lacking one or both copies of the NHE3 gene exhibited increased fluidity of their intestinal contents, which prevented the formation of obstructions and increased survival. Goblet cell hyperplasia was eliminated, but not the accumulation of Paneth cell granules or increased cell proliferation in the crypts. Microarray analysis of small intestine RNA from Cftr-null, NHE3-null, and double-null mice all revealed downregulation of genes involved in xenobiotic metabolism, including a cohort of genes involved in glutathione metabolism. Expression of energy metabolism genes was altered, but there were no changes in genes involved in inflammation. Total intracellular glutathione was increased in the jejunum of all of the mutants and the ratio of reduced to oxidized glutathione was reduced in Cftr-null mutants, indicating that CFTR deficiency affects intestinal glutathione metabolism. The data establish a major role for NHE3 in regulating the fluidity of the intestinal contents and show that reduced NHE3-mediated absorption reverses some of the intestinal pathologies of cystic fibrosis, thus suggesting that it may serve as a potential therapeutic target.
Figures
References
-
- Boucher RC Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med 13: 231–240, 2007. - PubMed
-
- Bruzzese E, Raia V, Gaudiello G, Polito G, Buccigrossi V, Formicola V, Guarino A. Intestinal inflammation is a frequent feature of cystic fibrosis and is reduced by probiotic administration. Aliment Pharmacol Ther 20: 813–819, 2004. - PubMed
-
- Childers M, Eckel G, Himmel A, Caldwell J. A new model of cystic fibrosis pathology: lack of transport of glutathione and its thiocyanate conjugates. Med Hypotheses 68: 101–112, 2007. - PubMed
-
- Clarke LL, Gawenis LR, Bradford EM, Judd LM, Boyle KT, Simpson JE, Shull GE, Tanabe H, Ouellette AJ, Franklin CL, Walker NM. Abnormal Paneth cell granule dissolution and compromised resistance to bacterial colonization in the intestine of CF mice. Am J Physiol Gastrointest Liver Physiol 286: G1050–G1058, 2004. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases