

Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism
- PMID: 19164857
- PMCID: PMC2631296
- DOI: 10.1172/JCI36376
Hedgehog signaling has a protective effect in glucocorticoid-induced mouse neonatal brain injury through an 11betaHSD2-dependent mechanism
Abstract
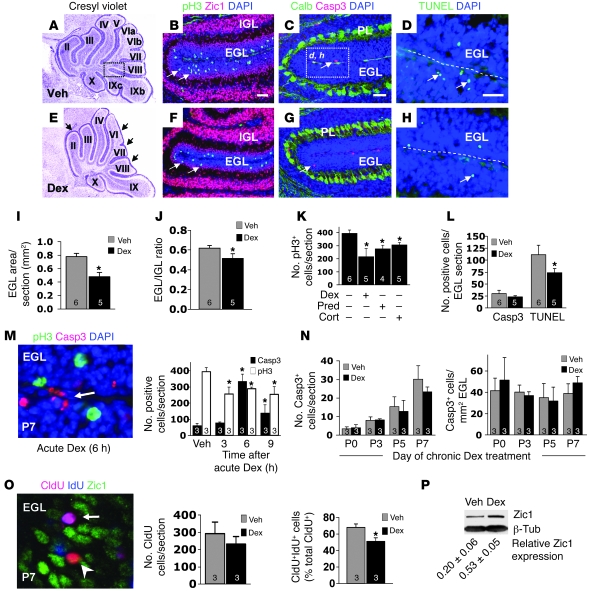
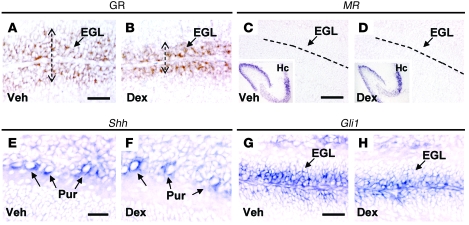
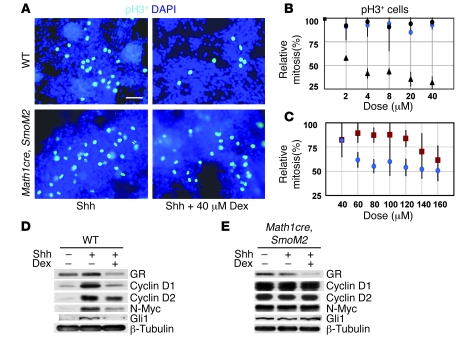
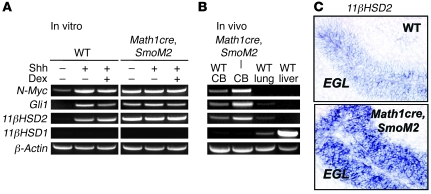
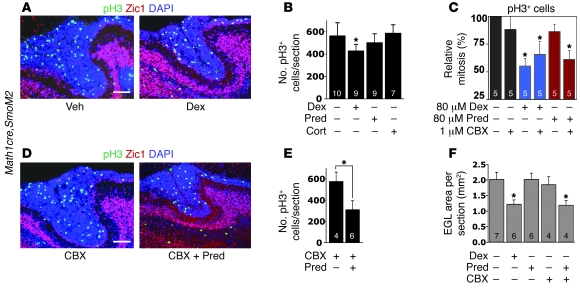
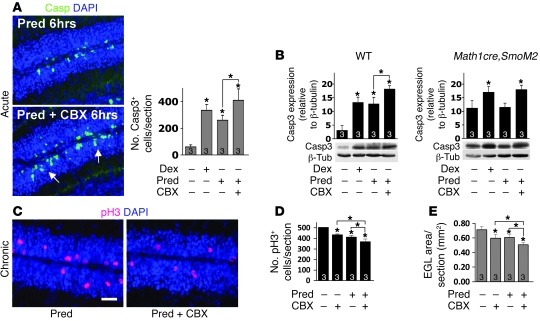
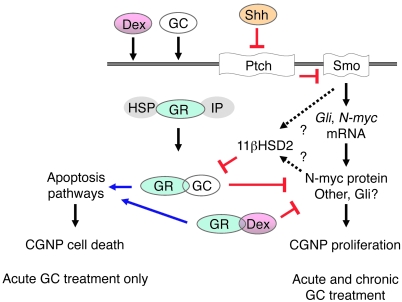
Glucocorticoids (GCs) are administered to human fetuses at risk of premature delivery and to infants with life-threatening respiratory and cardiac conditions. However, there are ongoing concerns about adverse effects of GC treatment on the developing human brain, although the precise molecular mechanisms underlying GC-induced brain injury are unclear. Here, we identified what we believe to be novel cross-antagonistic interactions of Sonic hedgehog (Shh) and GC signaling in proliferating mouse cerebellar granule neuron precursors (CGNPs). Chronic GC treatment (from P0 through P7) in mouse pups inhibited Shh-induced proliferation and upregulation of expression of N-myc, Gli1, and D-type cyclin protein in CGNPs. Conversely, acute GC treatment (on P7 only) caused transient apoptosis. Shh signaling antagonized these effects of GCs, in part by induction of 11beta-hydroxysteroid dehydrogenase type 2 (11betaHSD2). Importantly, 11betaHSD2 antagonized the effects of the GCs corticosterone, hydrocortisone, and prednisolone, but not the synthetic GC dexamethasone. Our findings indicate that Shh signaling is protective in the setting of GC-induced mouse neonatal brain injury. Furthermore, they led us to propose that 11betaHSD2-sensitive GCs (e.g., hydrocortisone) should be used in preference to dexamethasone in neonatal human infants because of the potential for reduced neurotoxicity.
Figures
Comment in
-
Glucocorticoids and neonatal brain injury: the hedgehog connection.J Clin Invest. 2009 Feb;119(2):243-6. doi: 10.1172/jci38387. J Clin Invest. 2009. PMID: 19244604 Free PMC article.
Similar articles
-
Glucocorticoids and neonatal brain injury: the hedgehog connection.J Clin Invest. 2009 Feb;119(2):243-6. doi: 10.1172/jci38387. J Clin Invest. 2009. PMID: 19244604 Free PMC article.
-
Sonic Hedgehog Agonist Protects Against Complex Neonatal Cerebellar Injury.Cerebellum. 2018 Apr;17(2):213-227. doi: 10.1007/s12311-017-0895-0. Cerebellum. 2018. PMID: 29134361 Free PMC article.
-
[Interaction of glucocorticoids and the Sonic Hedgehog pathway during brain development].Med Sci (Paris). 2009 Aug-Sep;25(8-9):713-7. doi: 10.1051/medsci/2009258-9713. Med Sci (Paris). 2009. PMID: 19765385 Review. French.
-
11beta-Hydroxysteroid dehydrogenase type 2 protects the neonatal cerebellum from deleterious effects of glucocorticoids.Neuroscience. 2006 Feb;137(3):865-73. doi: 10.1016/j.neuroscience.2005.09.037. Epub 2005 Nov 14. Neuroscience. 2006. PMID: 16289840 Free PMC article.
-
Evidence for adverse effect of perinatal glucocorticoid use on the developing brain.Korean J Pediatr. 2014 Mar;57(3):101-9. doi: 10.3345/kjp.2014.57.3.101. Epub 2014 Mar 31. Korean J Pediatr. 2014. PMID: 24778691 Free PMC article. Review.
Cited by
-
Bax deficiency prolongs cerebellar neurogenesis, accelerates medulloblastoma formation and paradoxically increases both malignancy and differentiation.Oncogene. 2013 May 2;32(18):2304-14. doi: 10.1038/onc.2012.248. Epub 2012 Jun 18. Oncogene. 2013. PMID: 22710714 Free PMC article.
-
Activation of Sonic Hedgehog Leads to Survival Enhancement of Astrocytes via the GRP78-Dependent Pathway in Mice Infected with Angiostrongylus cantonensis.Biomed Res Int. 2015;2015:674371. doi: 10.1155/2015/674371. Epub 2015 Apr 16. Biomed Res Int. 2015. PMID: 25961032 Free PMC article.
-
Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins.Nat Rev Mol Cell Biol. 2019 Mar;20(3):175-193. doi: 10.1038/s41580-018-0089-8. Nat Rev Mol Cell Biol. 2019. PMID: 30655609 Free PMC article. Review.
-
11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action.Physiol Rev. 2013 Jul;93(3):1139-206. doi: 10.1152/physrev.00020.2012. Physiol Rev. 2013. PMID: 23899562 Free PMC article. Review.
-
ECG in neonate mice with spinal muscular atrophy allows assessment of drug efficacy.Front Biosci (Elite Ed). 2015 Jan 1;7(1):107-16. doi: 10.2741/E721. Front Biosci (Elite Ed). 2015. PMID: 25553367 Free PMC article.
References
-
- Liggins G.C., Howie R.N. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
