

Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome
- PMID: 19164858
- PMCID: PMC2631294
- DOI: 10.1172/JCI35958
Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome
Abstract
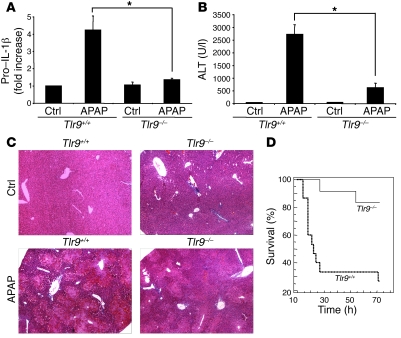
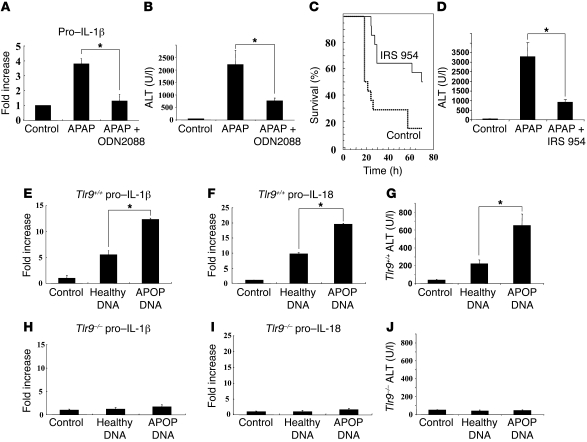
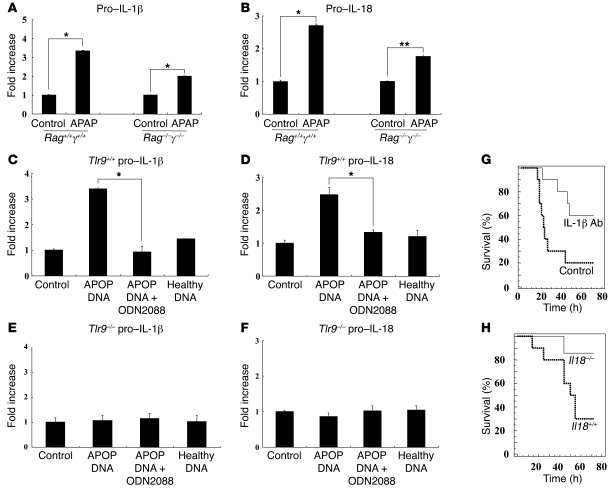
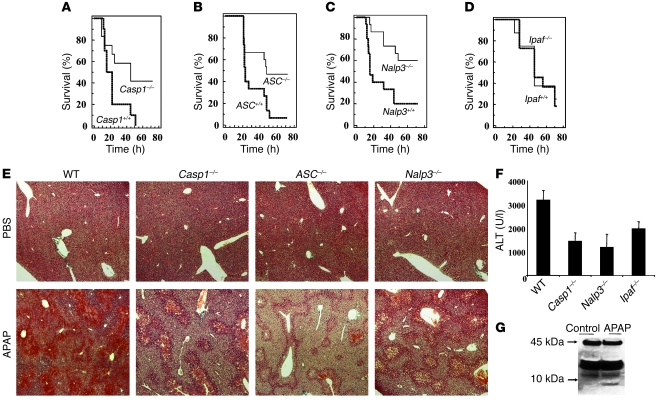
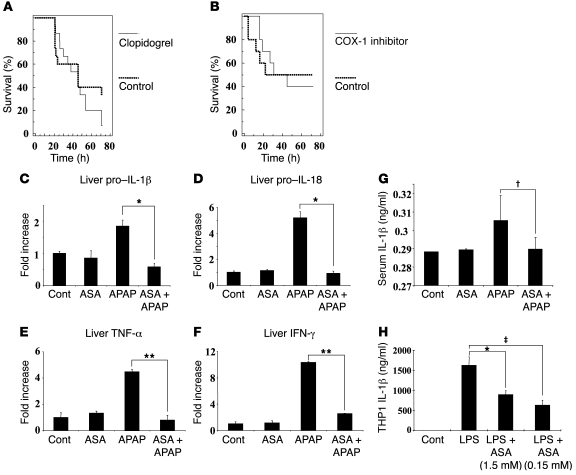
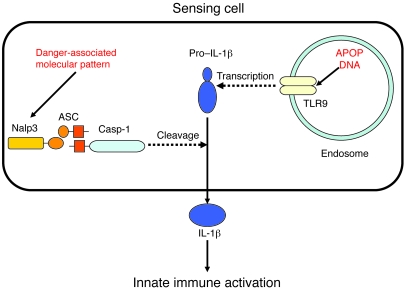
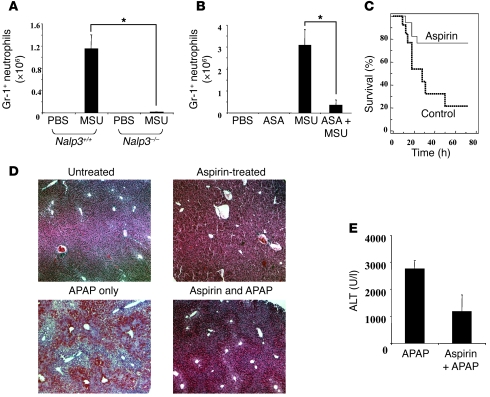
Hepatocyte death results in a sterile inflammatory response that amplifies the initial insult and increases overall tissue injury. One important example of this type of injury is acetaminophen-induced liver injury, in which the initial toxic injury is followed by innate immune activation. Using mice deficient in Tlr9 and the inflammasome components Nalp3 (NACHT, LRR, and pyrin domain-containing protein 3), ASC (apoptosis-associated speck-like protein containing a CARD), and caspase-1, we have identified a nonredundant role for Tlr9 and the Nalp3 inflammasome in acetaminophen-induced liver injury. We have shown that acetaminophen treatment results in hepatocyte death and that free DNA released from apoptotic hepatocytes activates Tlr9. This triggers a signaling cascade that increases transcription of the genes encoding pro-IL-1beta and pro-IL-18 in sinusoidal endothelial cells. By activating caspase-1, the enzyme responsible for generating mature IL-1beta and IL-18 from pro-IL-1beta and pro-IL-18, respectively, the Nalp3 inflammasome plays a crucial role in the second step of proinflammatory cytokine activation following acetaminophen-induced liver injury. Tlr9 antagonists and aspirin reduced mortality from acetaminophen hepatotoxicity. The protective effect of aspirin on acetaminophen-induced liver injury was due to downregulation of proinflammatory cytokines, rather than inhibition of platelet degranulation or COX-1 inhibition. In summary, we have identified a 2-signal requirement (Tlr9 and the Nalp3 inflammasome) for acetaminophen-induced hepatotoxicity and some potential therapeutic approaches.
Figures
Comment in
-
DAMPs ramp up drug toxicity.J Clin Invest. 2009 Feb;119(2):246-9. doi: 10.1172/jci38178. J Clin Invest. 2009. PMID: 19244605 Free PMC article.
-
Inflammasome and toll-like receptor 9: Partners in crime in toxic liver injury.Hepatology. 2009 Jun;49(6):2119-21. doi: 10.1002/hep.23022. Hepatology. 2009. PMID: 19479796 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
