

A structural overview of the vertebrate prion proteins
- PMID: 19164911
- PMCID: PMC2634592
- DOI: 10.4161/pri.1.3.5281
A structural overview of the vertebrate prion proteins
Abstract
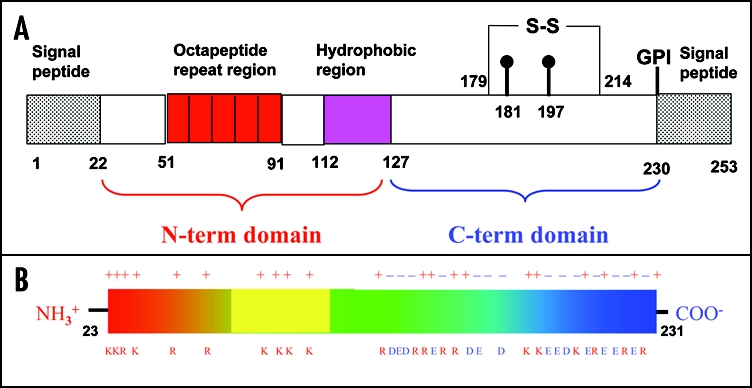
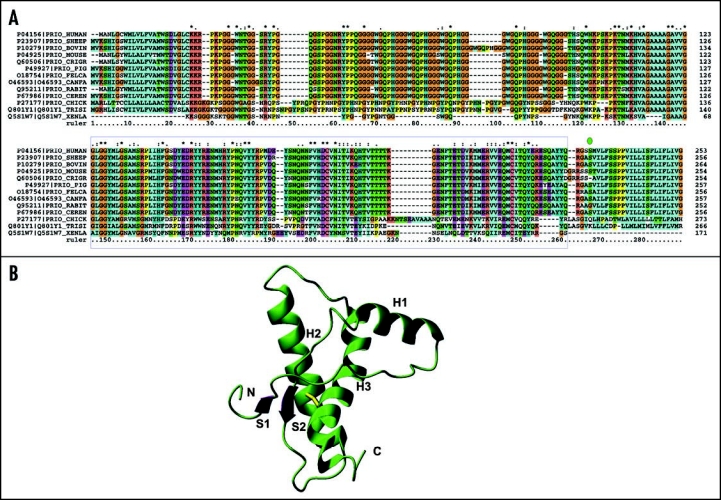
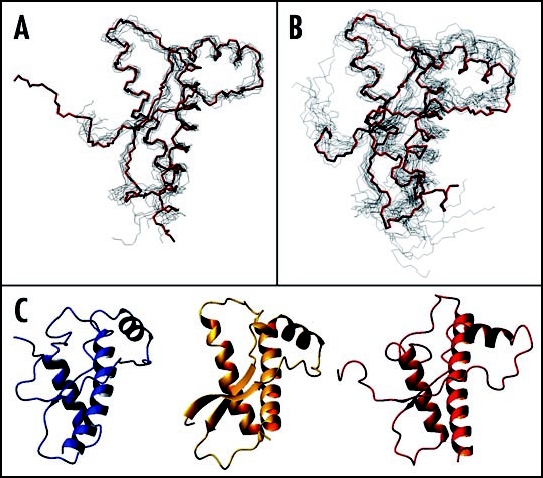
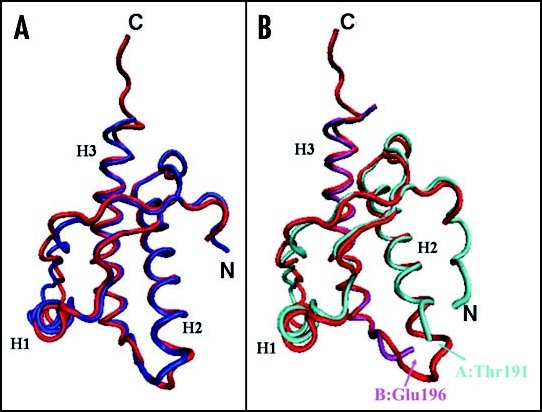
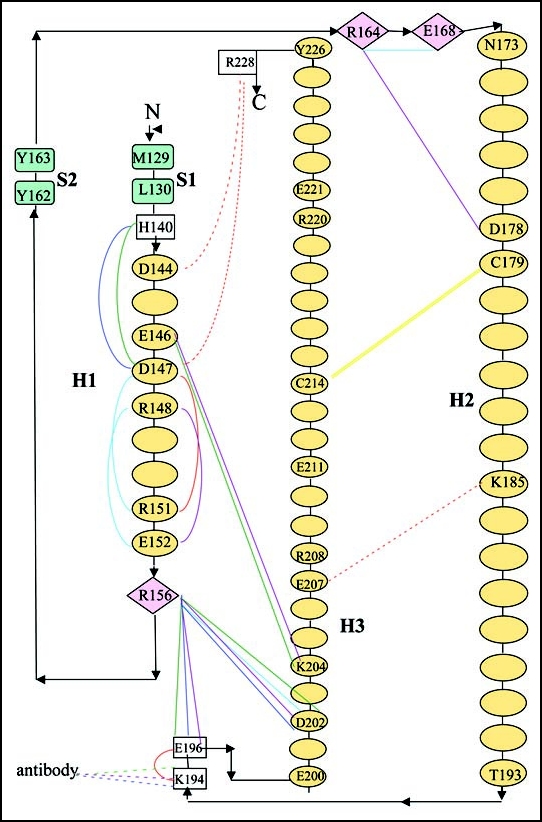
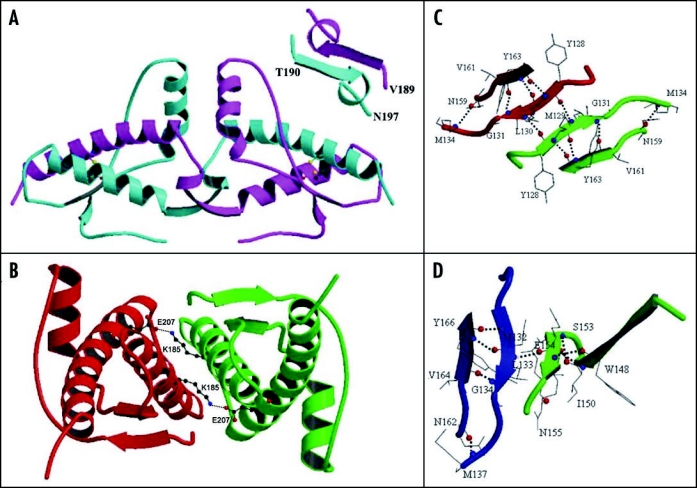
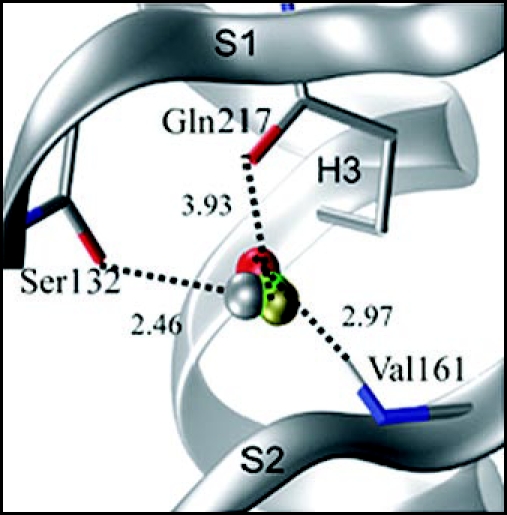
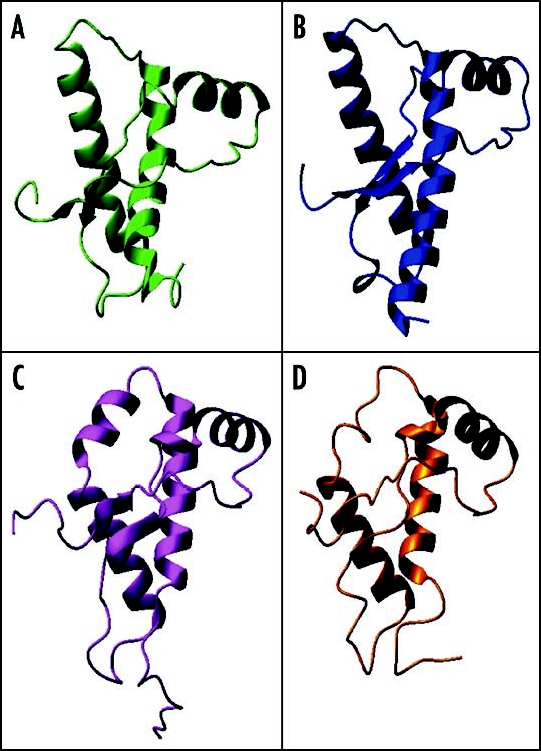
Among the diseases caused by protein misfolding is the family associated with the prion protein (PrP). This is a small extracellular membrane-anchored molecule of yet unknown function. Understanding how PrP folds both into its cellular and pathological forms is thought to be crucial for explaining protein misfolding in general and the specific role of PrP in disease. Since the first structure determination, an increasing number of structural studies of PrP have become available, showing that the protein is formed by a flexible N-terminal region and a highly conserved globular C-terminal domain. We review here the current knowledge on PrP structure. We focus on vertebrate PrPs and analyse in detail the similarities and the differences among the coordinates of the C-terminal domain of PrP from different species, in search for understanding the mechanism of disease-causing mutations and the molecular bases of species barrier.
Figures
References
-
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. - PubMed
-
- Prusiner SB. Prions are novel infectious pathogens causing scrapie and Creutzfeldt-Jakob disease. Bioessays. 1986;5:281–286. - PubMed
-
- Prusiner SB. Human prion diseases and neurodegeneration. Curr Top Microbiol Immunol. 1996;207:1–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials