

Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics
- PMID: 19167289
- PMCID: PMC2716522
- DOI: 10.1016/j.bpj.2008.09.041
Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics
Abstract
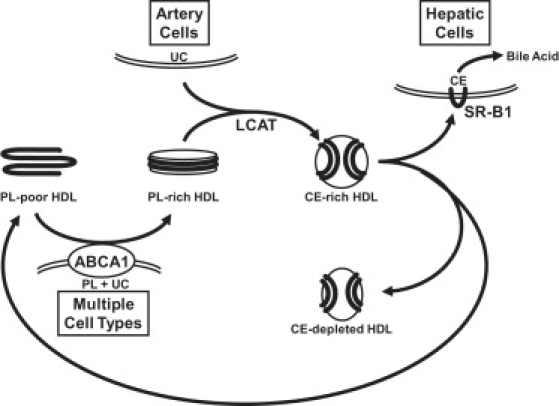
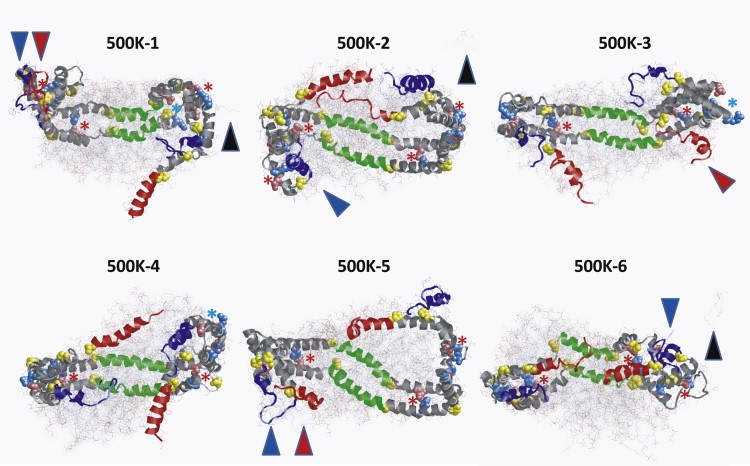
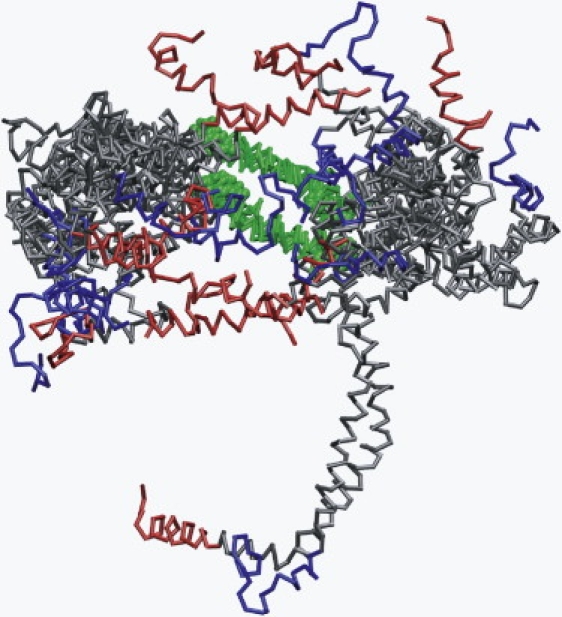
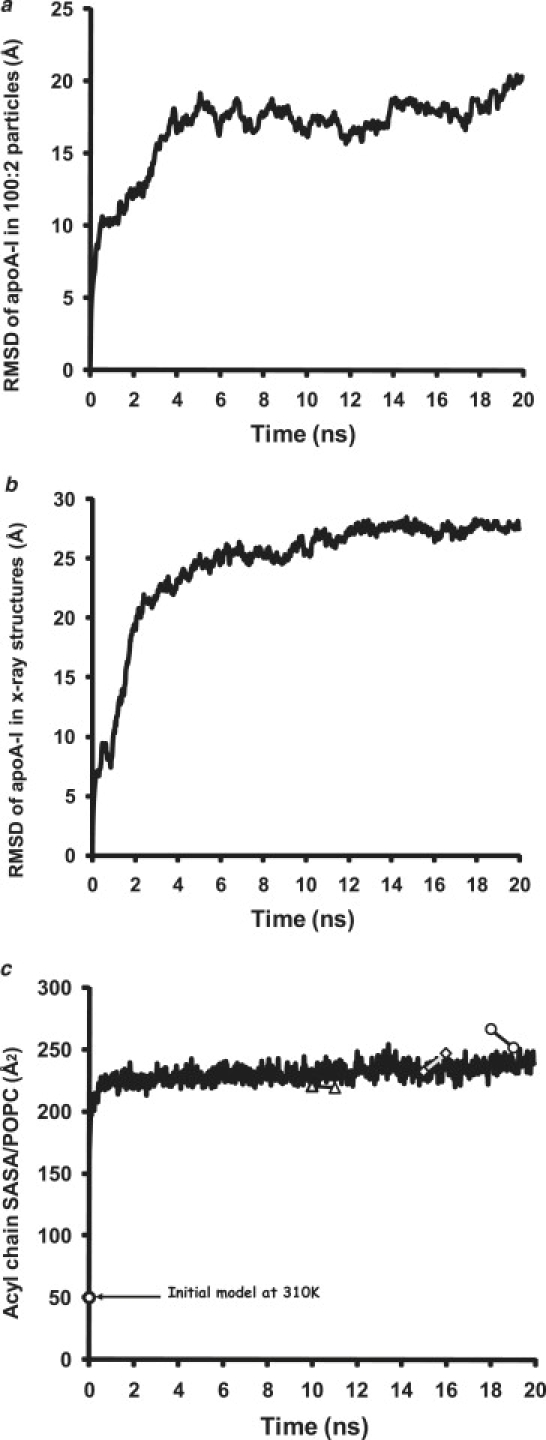
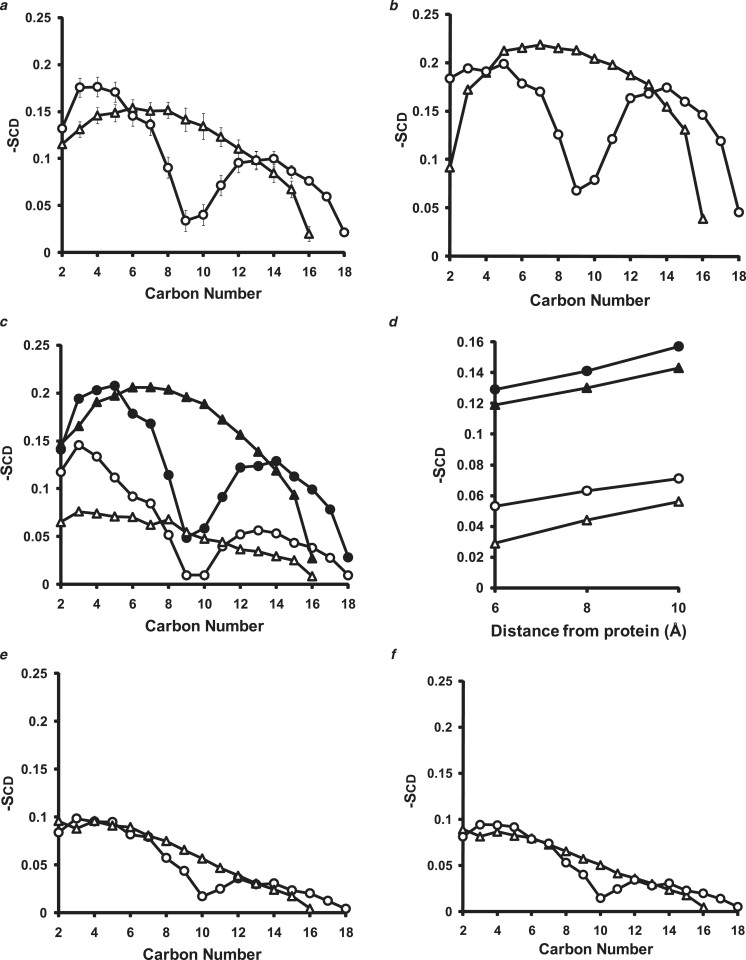
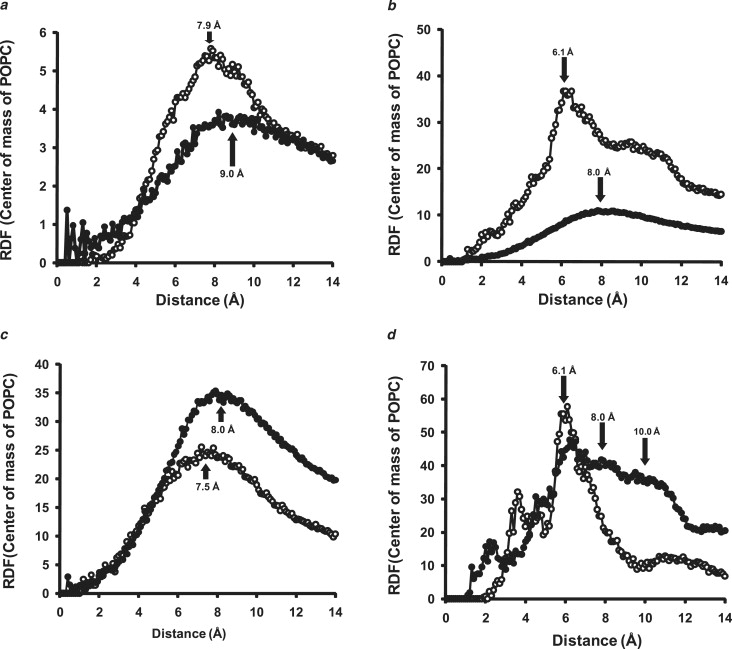
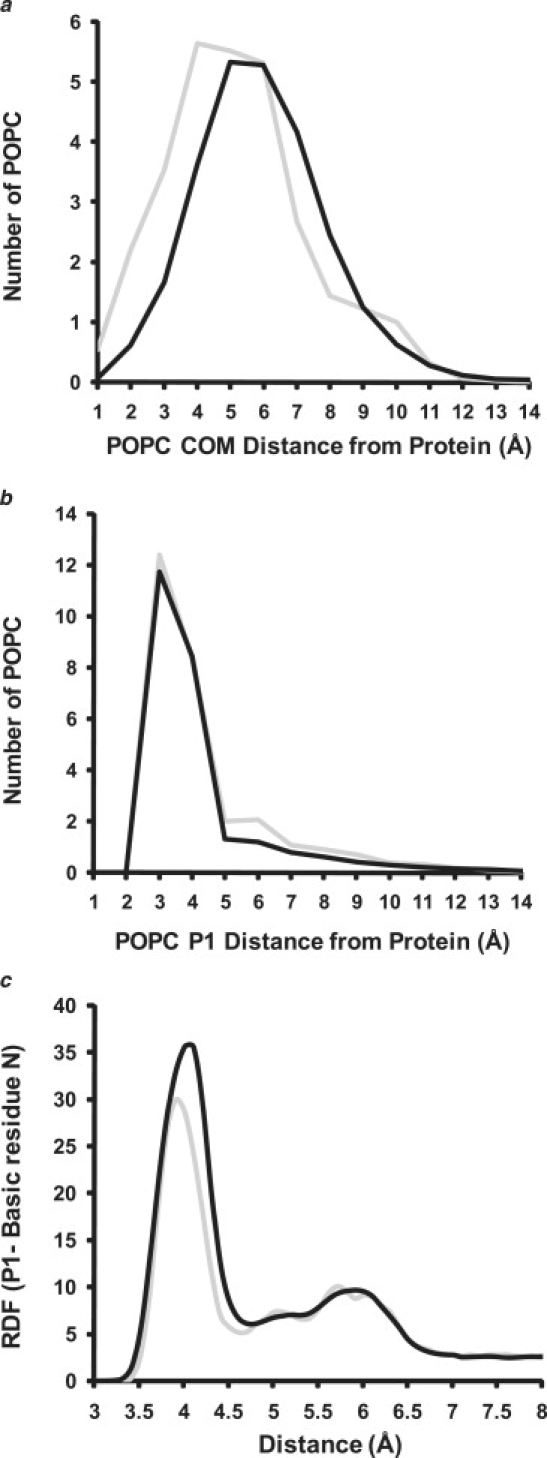
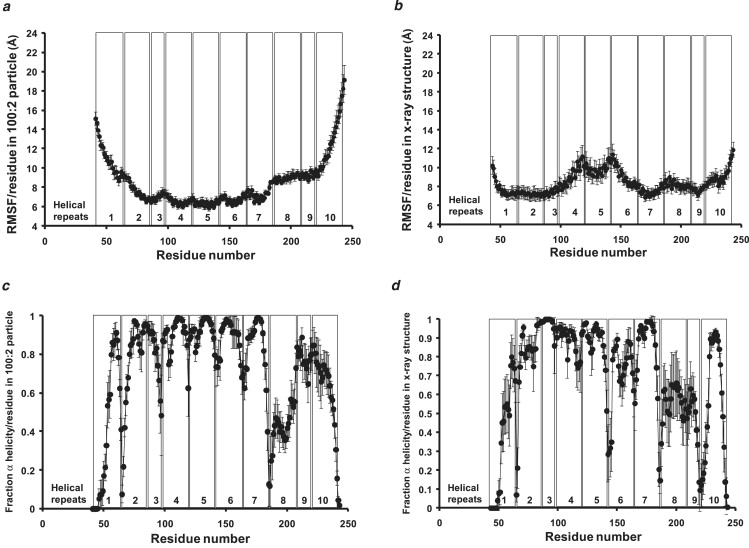
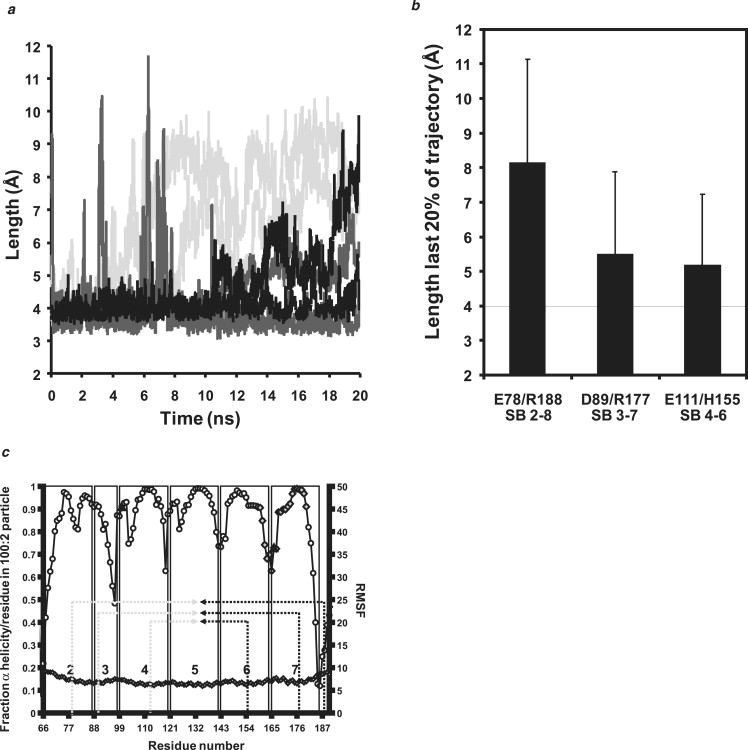
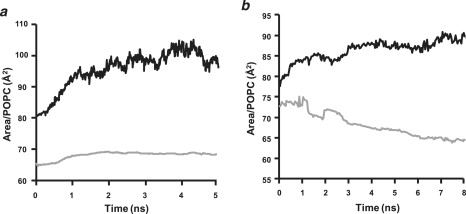
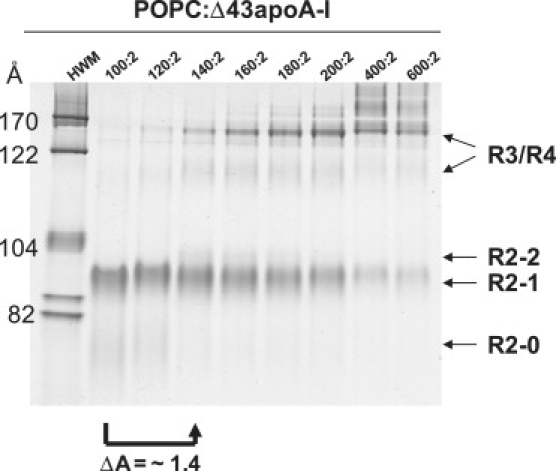
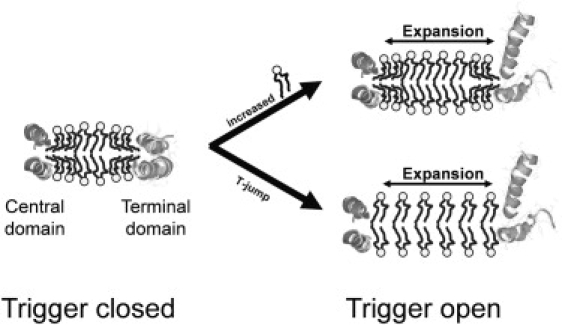
Apolipoprotein (apo) A-I is an unusually flexible protein whose lipid-associated structure is poorly understood. Thermal denaturation, which is used to measure the global helix stability of high-density lipoprotein (HDL)-associated apoA-I, provides no information about local helix stability. Here we report the use of temperature jump molecular dynamics (MD) simulations to scan the per-residue helix stability of apoA-I in phospholipid-rich HDL. When three 20 ns MD simulations were performed at 500 K on each of two particles created by MD simulations at 310 K, bilayers remained intact but expanded by 40%, and total apoA-I helicity decreased from 95% to 72%. Of significance, the conformations of the overlapping N- and C-terminal domains of apoA-I in the particles were unusually mobile, exposing hydrocarbon regions of the phospholipid to solvent; a lack of buried interhelical salt bridges in the terminal domains correlated with increased mobility. Nondenaturing gradient gels show that 40% expansion of the phospholipid surface of 100:2 particles by addition of palmitoyloleoylphosphatidylcholine exceeds the threshold of particle stability. As a unifying hypothesis, we propose that the terminal domains of apoA-I are phospholipid concentration-sensitive molecular triggers for fusion/remodeling of HDL particles. Since HDL remodeling is necessary for cholesterol transport, our model for remodeling has substantial biomedical implications.
Figures
References
-
- Linsel-Nitschke P., Tall A.R. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat. Rev. Drug Discov. 2005;4:193–205. - PubMed
-
- Oram J.F., Heinecke J.W. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol. Rev. 2005;85:1343–1372. - PubMed
-
- Nichols A.V., Blanche P.J., Gong E.L., Shore V.G., Forte T.M. Molecular pathways in the transformation of model discoidal lipoprotein complexes induced by lecithin:cholesterol acyltransferase. Biochim. Biophys. Acta. 1985;834:285–300. - PubMed
-
- Trigatti B.L., Krieger M., Rigotti A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003;23:1732–1738. - PubMed
-
- Rye K.A., Clay M.A., Barter P.J. Remodelling of high density lipoproteins by plasma factors. Atherosclerosis. 1999;145:227–238. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
