

Induction of hepatitis by JNK-mediated expression of TNF-alpha
- PMID: 19167327
- PMCID: PMC2794880
- DOI: 10.1016/j.cell.2008.11.017
Induction of hepatitis by JNK-mediated expression of TNF-alpha
Abstract
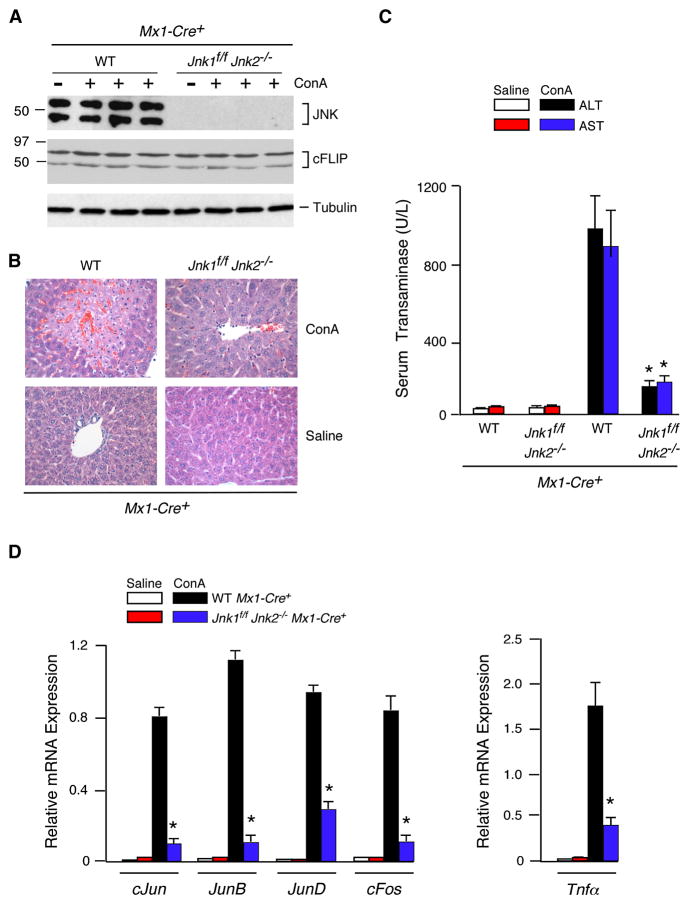
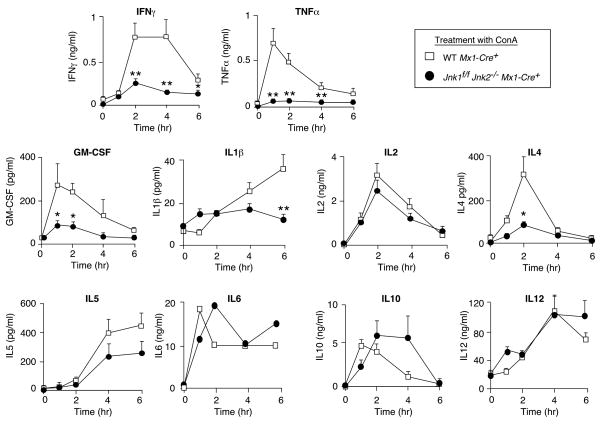
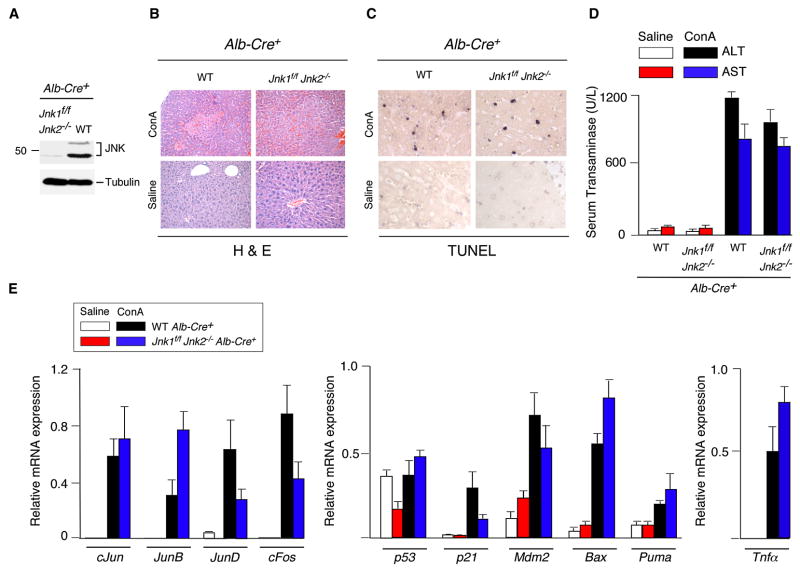
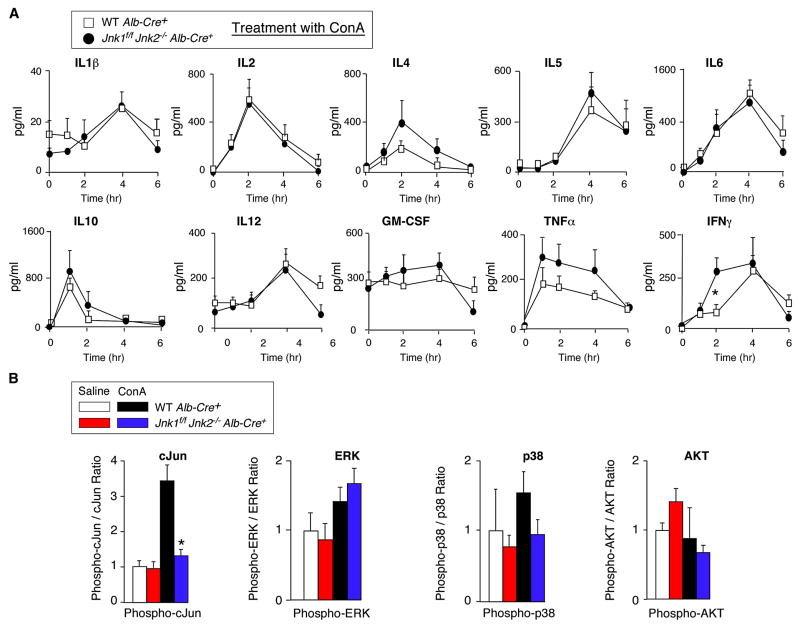
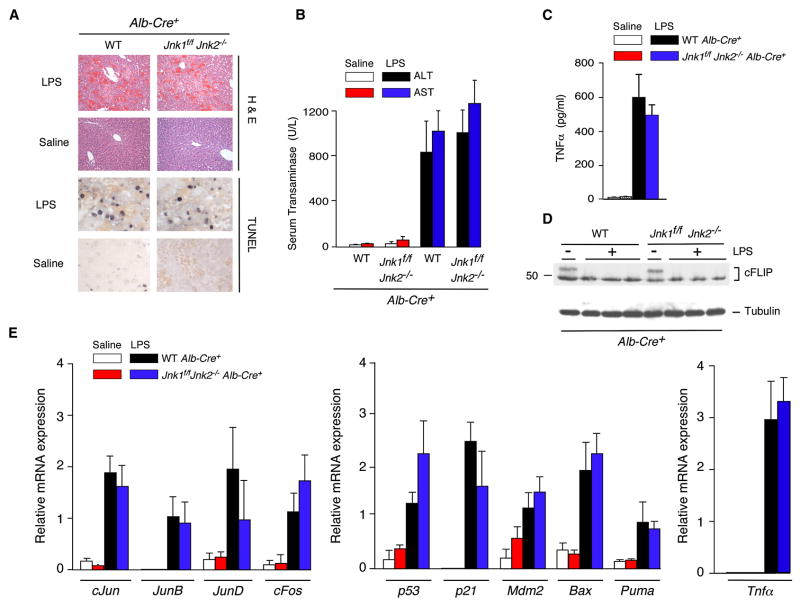
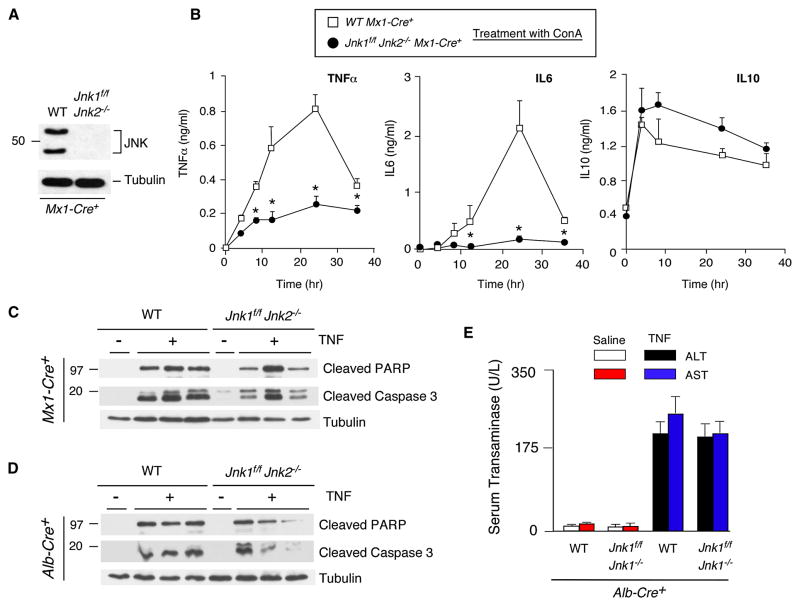
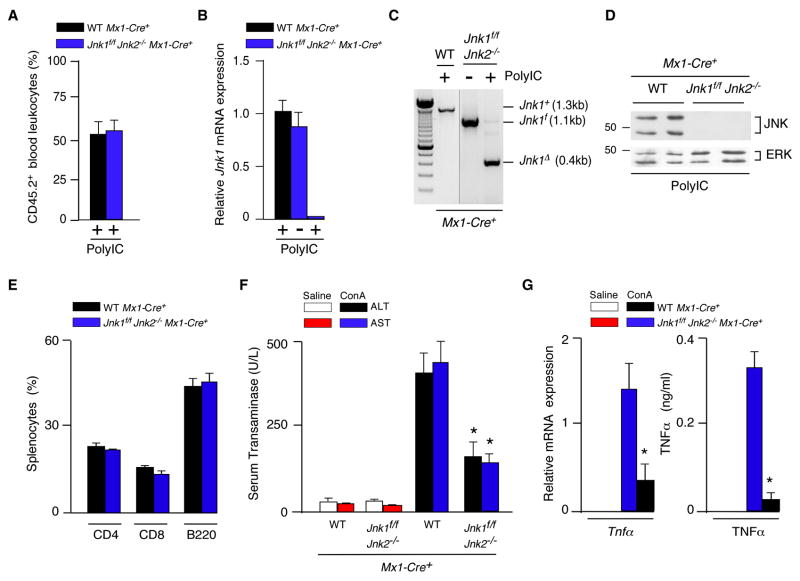
The c-Jun NH(2)-terminal kinase (JNK) signaling pathway has been implicated in the development of tumor necrosis factor (TNF)-dependent hepatitis. JNK may play a critical role in hepatocytes during TNF-stimulated cell death in vivo. To test this hypothesis, we examined the phenotype of mice with compound disruption of the Jnk1 and Jnk2 genes. Mice with loss of JNK1/2 expression in hepatocytes exhibited no defects in the development of hepatitis compared with control mice, whereas mice with loss of JNK1/2 in the hematopoietic compartment exhibited a profound defect in hepatitis that was associated with markedly reduced expression of TNF-alpha. These data indicate that JNK is required for TNF-alpha expression but not for TNF-alpha-stimulated death of hepatocytes. Indeed, TNF-alpha induced similar hepatic damage in both mice with hepatocyte-specific JNK1/2 deficiency and control mice. These observations confirm a role for JNK in the development of hepatitis but identify hematopoietic cells as the site of the essential function of JNK.
Figures
References
-
- Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. - PubMed
-
- Alves-Guerra MC, Rousset S, Pecqueur C, Mallat Z, Blanc J, Tedgui A, Bouillaud F, Cassard-Doulcier AM, Ricquier D, Miroux B. Bone marrow transplantation reveals the in vivo expression of the mitochondrial uncoupling protein 2 in immune and nonimmune cells during inflammation. J Biol Chem. 2003;278:42307–42312. - PubMed
-
- Bonder CS, Ajuebor MN, Zbytnuik LD, Kubes P, Swain MG. Essential role for neutrophil recruitment to the liver in concanavalin A-induced hepatitis. J Immunol. 2004;172:45–53. - PubMed
-
- Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30:652–660. - PubMed
-
- Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
