

Antibody to the dendritic cell surface activation antigen CD83 prevents acute graft-versus-host disease
- PMID: 19171763
- PMCID: PMC2646577
- DOI: 10.1084/jem.20070723
Antibody to the dendritic cell surface activation antigen CD83 prevents acute graft-versus-host disease
Erratum in
- J Exp Med. 2009 May 11;206(5):1203
Abstract
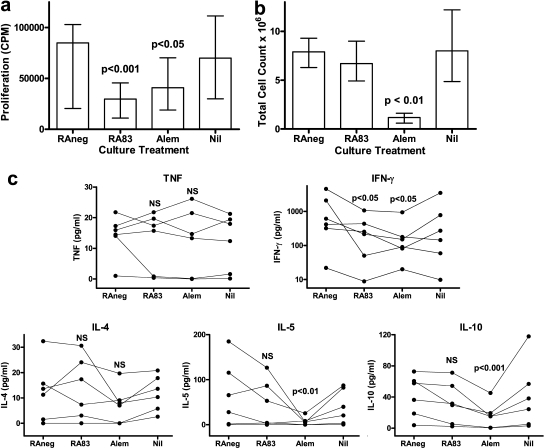
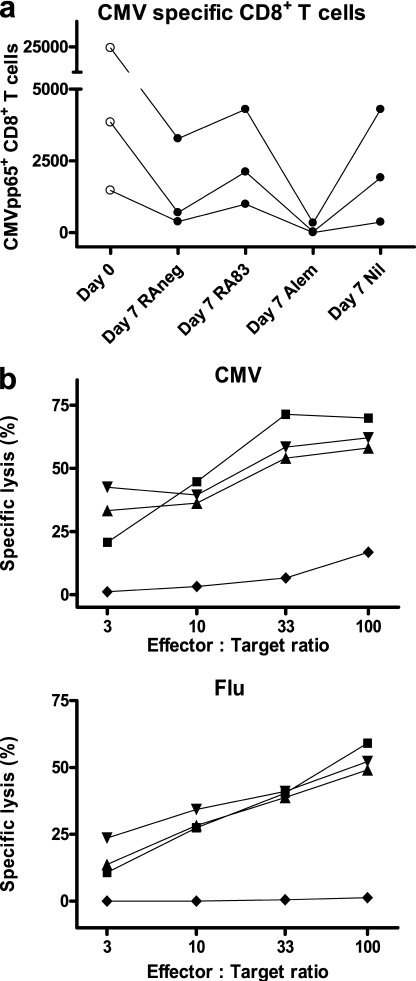
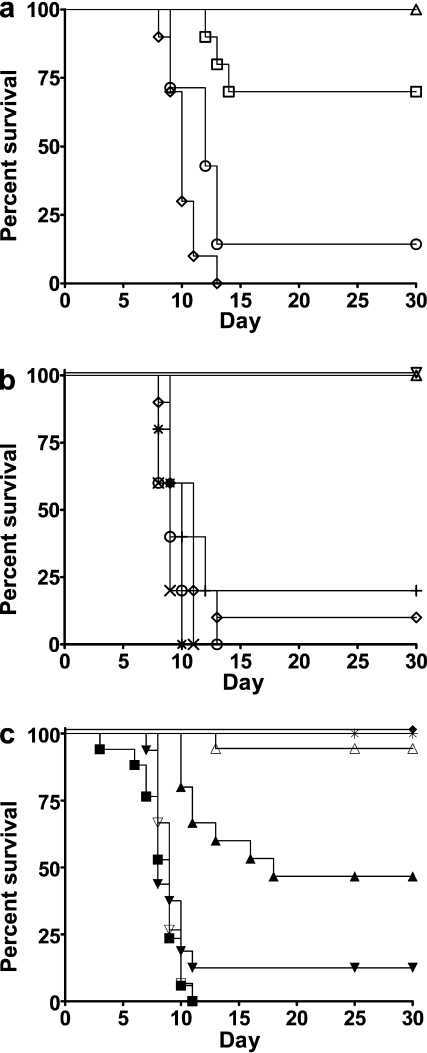
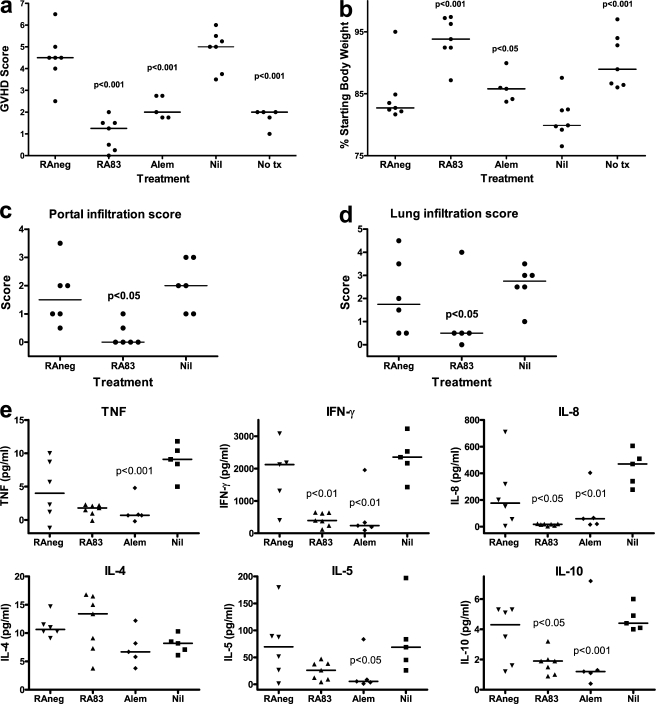
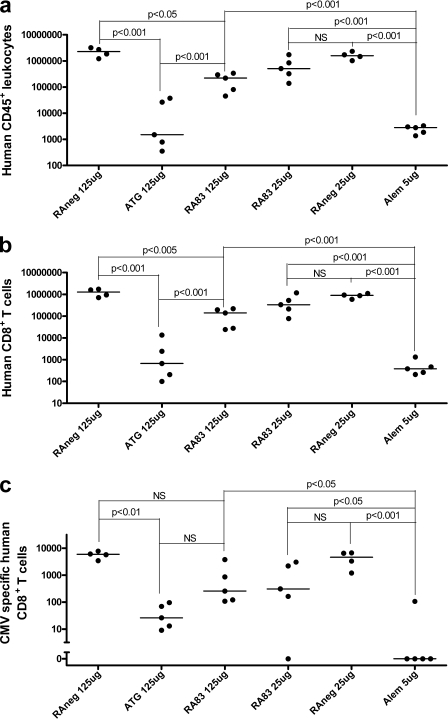
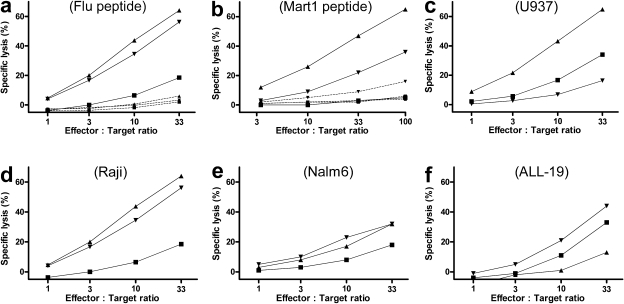
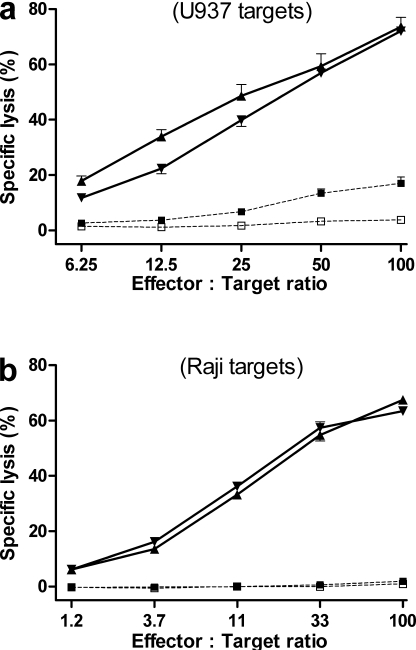
Allogeneic (allo) hematopoietic stem cell transplantation is an effective therapy for hematological malignancies but it is limited by acute graft-versus-host disease (GVHD). Dendritic cells (DC) play a major role in the allo T cell stimulation causing GVHD. Current immunosuppressive measures to control GVHD target T cells but compromise posttransplant immunity in the patient, particularly to cytomegalovirus (CMV) and residual malignant cells. We showed that treatment of allo mixed lymphocyte cultures with activated human DC-depleting CD83 antibody suppressed alloproliferation but preserved T cell numbers, including those specific for CMV. We also tested CD83 antibody in the human T cell-dependent peripheral blood mononuclear cell transplanted SCID (hu-SCID) mouse model of GVHD. We showed that this model requires human DC and that CD83 antibody treatment prevented GVHD but, unlike conventional immunosuppressants, did not prevent engraftment of human T cells, including cytotoxic T lymphocytes (CTL) responsive to viruses and malignant cells. Immunization of CD83 antibody-treated hu-SCID mice with irradiated human leukemic cell lines induced allo antileukemic CTL effectors in vivo that lysed (51)Cr-labeled leukemic target cells in vitro without further stimulation. Antibodies that target activated DC are a promising new therapeutic approach to the control of GVHD.
Figures
References
-
- Welniak L.A., Blazar B.R., Murphy W.J. 2007. Immunobiology of allogeneic hematopoietic stem cell transplantation.Annu. Rev. Immunol. 25:139–170 - PubMed
-
- Shlomchik W.D., Couzens M.S., Tang C.B., McNiff J., Robert M.E., Liu J., Shlomchik M.J., Emerson S.G. 1999. Prevention of graft versus host disease by inactivation of host antigen-presenting cells.Science. 285:412–415 - PubMed
-
- Duffner U.A., Maeda Y., Cooke K.R., Reddy P., Ordemann R., Liu C., Ferrara J.L., Teshima T. 2004. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease.J. Immunol. 172:7393–7398 - PubMed
-
- Matte C.C., Liu J., Cormier J., Anderson B.E., Athanasiadis I., Jain D., McNiff J., Shlomchik W.D. 2004. Donor APCs are required for maximal GVHD but not for GVL.Nat. Med. 10:987–992 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
