

Combinatorial effects of environmental parameters on transcriptional regulation in Saccharomyces cerevisiae: a quantitative analysis of a compendium of chemostat-based transcriptome data
- PMID: 19173729
- PMCID: PMC2640415
- DOI: 10.1186/1471-2164-10-53
Combinatorial effects of environmental parameters on transcriptional regulation in Saccharomyces cerevisiae: a quantitative analysis of a compendium of chemostat-based transcriptome data
Abstract
Background: Microorganisms adapt their transcriptome by integrating multiple chemical and physical signals from their environment. Shake-flask cultivation does not allow precise manipulation of individual culture parameters and therefore precludes a quantitative analysis of the (combinatorial) influence of these parameters on transcriptional regulation. Steady-state chemostat cultures, which do enable accurate control, measurement and manipulation of individual cultivation parameters (e.g. specific growth rate, temperature, identity of the growth-limiting nutrient) appear to provide a promising experimental platform for such a combinatorial analysis.
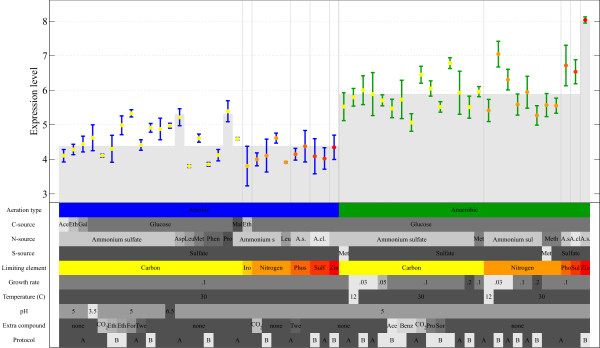
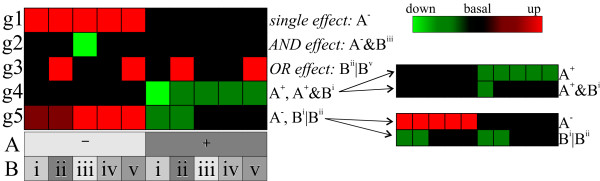
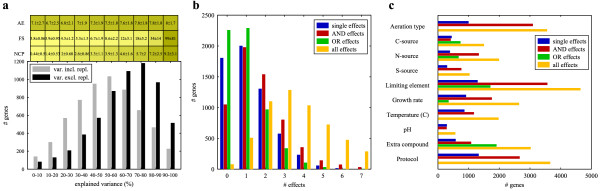
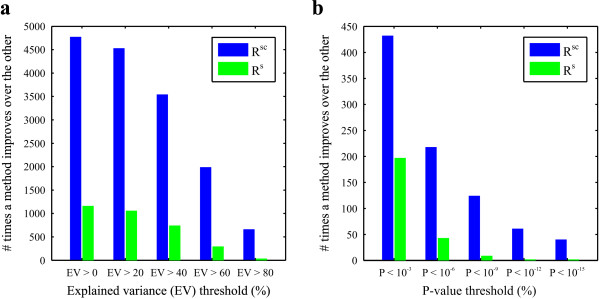
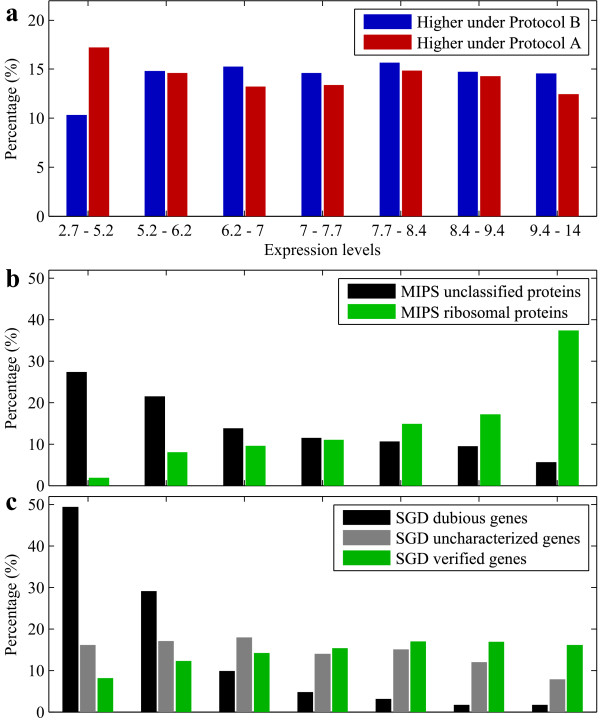
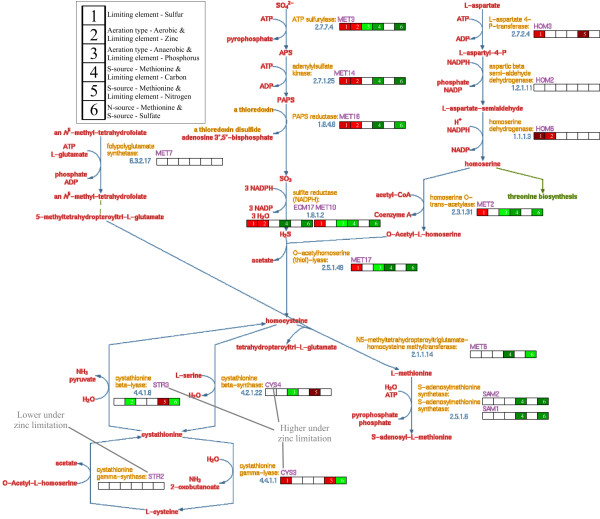
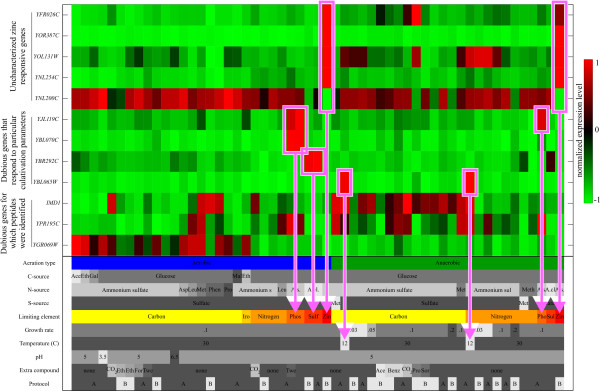
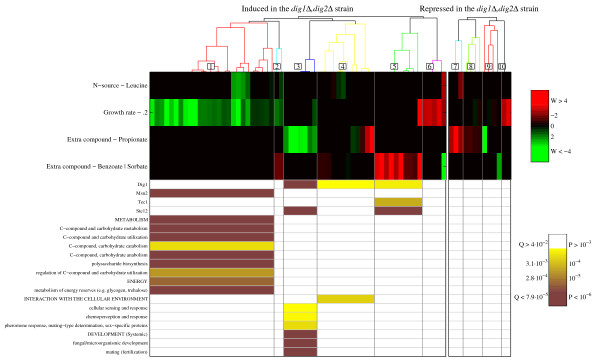
Results: A microarray compendium of 170 steady-state chemostat cultures of the yeast Saccharomyces cerevisiae is presented and analyzed. The 170 microarrays encompass 55 unique conditions, which can be characterized by the combined settings of 10 different cultivation parameters. By applying a regression model to assess the impact of (combinations of) cultivation parameters on the transcriptome, most S. cerevisiae genes were shown to be influenced by multiple cultivation parameters, and in many cases by combinatorial effects of cultivation parameters. The inclusion of these combinatorial effects in the regression model led to higher explained variance of the gene expression patterns and resulted in higher function enrichment in subsequent analysis. We further demonstrate the usefulness of the compendium and regression analysis for interpretation of shake-flask-based transcriptome studies and for guiding functional analysis of (uncharacterized) genes and pathways.
Conclusion: Modeling the combinatorial effects of environmental parameters on the transcriptome is crucial for understanding transcriptional regulation. Chemostat cultivation offers a powerful tool for such an approach.
Figures

References
-
- Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, Cunningham ML, Deng S, Dressman HK, Fannin RD, Farin FM, Freedman JH, Fry RC, Harper A, Humble MC, Hurban P, Kavanagh TJ, Kaufmann WK, Kerr KF, Jing L, Lapidus JA, Lasarev MR, Li J, Li YJ, Lobenhofer EK, Lu X, Malek RL, Milton S, Nagalla SR, O'malley JP, Palmer VS, Pattee P, Paules RS, Perou CM, Phillips K, Qin LX, Qiu Y, Quigley SD, Rodland M, Rusyn I, Samson LD, Schwartz DA, Shi Y, Shin JL, Sieber SO, Slifer S, Speer MC, Spencer PS, Sproles DI, Swenberg JA, Suk WA, Sullivan RC, Tian R, Tennant RW, Todd SA, Tucker CJ, Houten BV, Weis BK, Xuan S, Zarbl H, of the Toxicogenomics Research Consortium M Standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth754. - DOI - PubMed
-
- Tai SL, Boer VM, Daran-Lapujade P, Walsh MC, de Winde JH, Daran JM, Pronk JT. Two-dimensional transcriptome analysis in chemostat cultures. Combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J Biol Chem. 2005;280:437–447. doi: 10.1074/jbc.M501243200. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
