

The Genome Reverse Compiler: an explorative annotation tool
- PMID: 19173744
- PMCID: PMC2640359
- DOI: 10.1186/1471-2105-10-35
The Genome Reverse Compiler: an explorative annotation tool
Abstract
Background: As sequencing costs have decreased, whole genome sequencing has become a viable and integral part of biological laboratory research. However, the tools with which genes can be found and functionally characterized have not been readily adapted to be part of the everyday biological sciences toolkit. Most annotation pipelines remain as a service provided by large institutions or come as an unwieldy conglomerate of independent components, each requiring their own setup and maintenance.
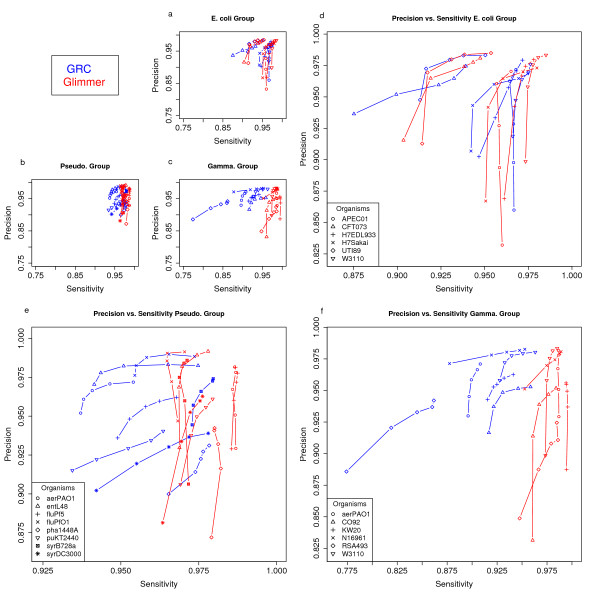
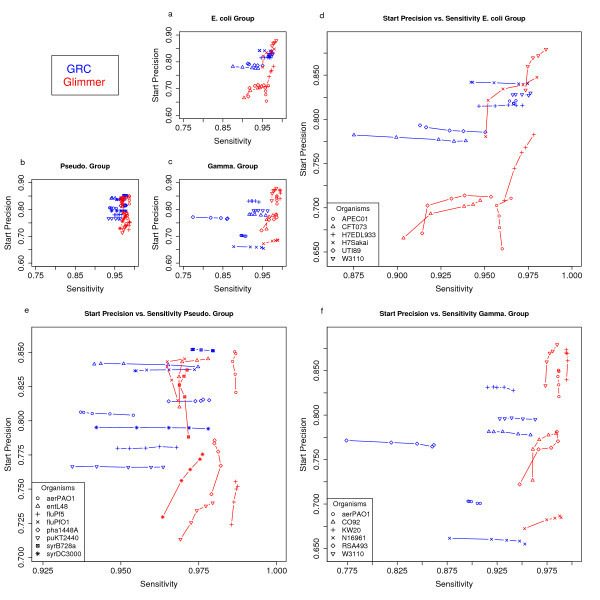
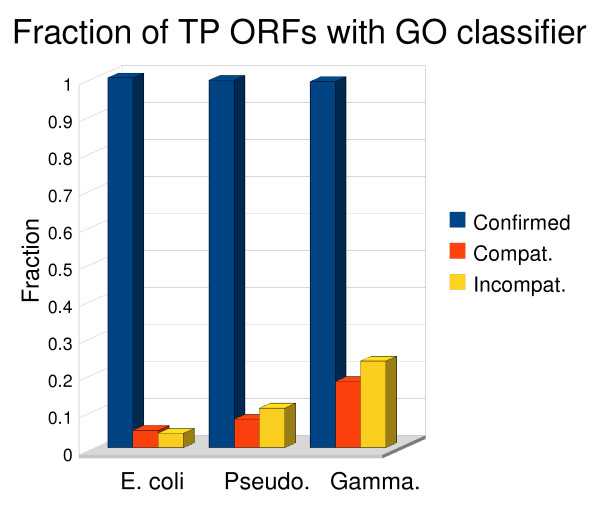
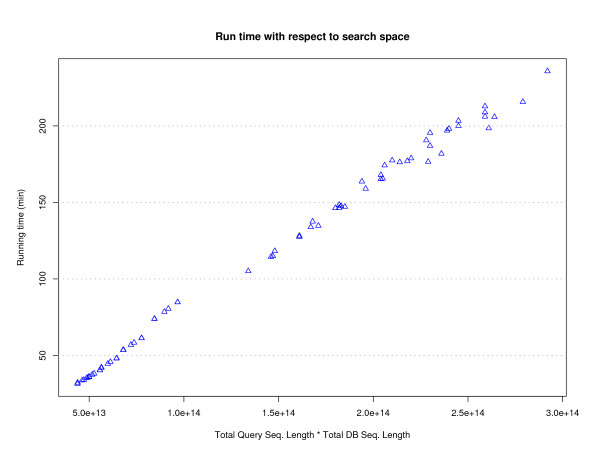
Results: To address this issue we have created the Genome Reverse Compiler, an easy-to-use, open-source, automated annotation tool. The GRC is independent of third party software installs and only requires a Linux operating system. This stands in contrast to most annotation packages, which typically require installation of relational databases, sequence similarity software, and a number of other programming language modules. We provide details on the methodology used by GRC and evaluate its performance on several groups of prokaryotes using GRC's built in comparison module.
Conclusion: Traditionally, to perform whole genome annotation a user would either set up a pipeline or take advantage of an online service. With GRC the user need only provide the genome he or she wants to annotate and the function resource files to use. The result is high usability and a very minimal learning curve for the intended audience of life science researchers and bioinformaticians. We believe that the GRC fills a valuable niche in allowing users to perform explorative, whole-genome annotation.
Figures
Similar articles
-
Next generation models for storage and representation of microbial biological annotation.BMC Bioinformatics. 2010 Oct 7;11 Suppl 6(Suppl 6):S15. doi: 10.1186/1471-2105-11-S6-S15. BMC Bioinformatics. 2010. PMID: 20946598 Free PMC article.
-
JUICE: a data management system that facilitates the analysis of large volumes of information in an EST project workflow.BMC Bioinformatics. 2006 Nov 23;7:513. doi: 10.1186/1471-2105-7-513. BMC Bioinformatics. 2006. PMID: 17123449 Free PMC article.
-
GeneTools--application for functional annotation and statistical hypothesis testing.BMC Bioinformatics. 2006 Oct 24;7:470. doi: 10.1186/1471-2105-7-470. BMC Bioinformatics. 2006. PMID: 17062145 Free PMC article.
-
VEGA, the genome browser with a difference.Brief Bioinform. 2005 Jun;6(2):189-93. doi: 10.1093/bib/6.2.189. Brief Bioinform. 2005. PMID: 15975227 Review.
-
Programming cells: towards an automated 'Genetic Compiler'.Curr Opin Biotechnol. 2010 Aug;21(4):572-81. doi: 10.1016/j.copbio.2010.07.005. Epub 2010 Aug 9. Curr Opin Biotechnol. 2010. PMID: 20702081 Free PMC article. Review.
Cited by
-
Draft Genome Sequence of FT9, a Novel Bacillus cereus Strain Isolated from a Brazilian Thermal Spring.Genome Announc. 2014 Oct 9;2(5):e01027-14. doi: 10.1128/genomeA.01027-14. Genome Announc. 2014. PMID: 25301660 Free PMC article.
-
CNN-MGP: Convolutional Neural Networks for Metagenomics Gene Prediction.Interdiscip Sci. 2019 Dec;11(4):628-635. doi: 10.1007/s12539-018-0313-4. Epub 2018 Dec 27. Interdiscip Sci. 2019. PMID: 30588558 Free PMC article.
-
Complete sequencing of Novosphingobium sp. PP1Y reveals a biotechnologically meaningful metabolic pattern.BMC Genomics. 2014 May 19;15(1):384. doi: 10.1186/1471-2164-15-384. BMC Genomics. 2014. PMID: 24884518 Free PMC article.
-
Patterns and processes of Mycobacterium bovis evolution revealed by phylogenomic analyses.Genome Biol Evol. 2017 Feb 13;9(3):521-35. doi: 10.1093/gbe/evx022. Online ahead of print. Genome Biol Evol. 2017. PMID: 28201585 Free PMC article.
-
Complete genome sequence of Mycobacterium massiliense.J Bacteriol. 2012 Oct;194(19):5455. doi: 10.1128/JB.01219-12. J Bacteriol. 2012. PMID: 22965084 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
