

The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals
- PMID: 19174508
- PMCID: PMC2644154
- DOI: 10.1073/pnas.0810934106
The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals
Abstract
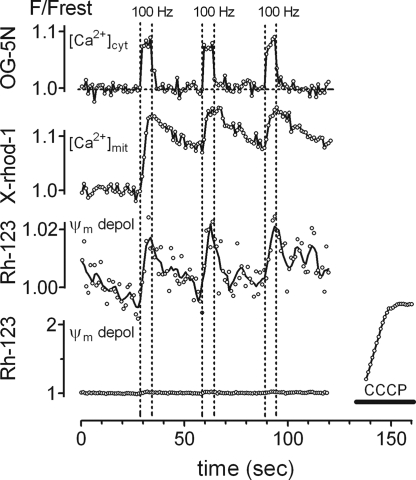
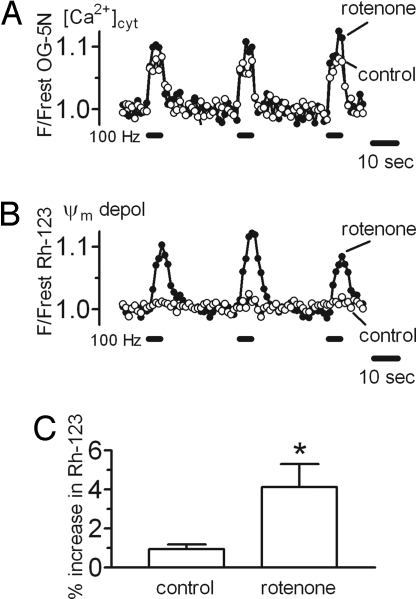
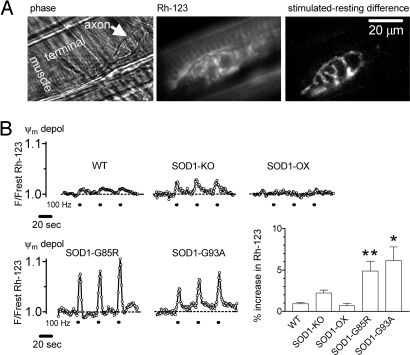
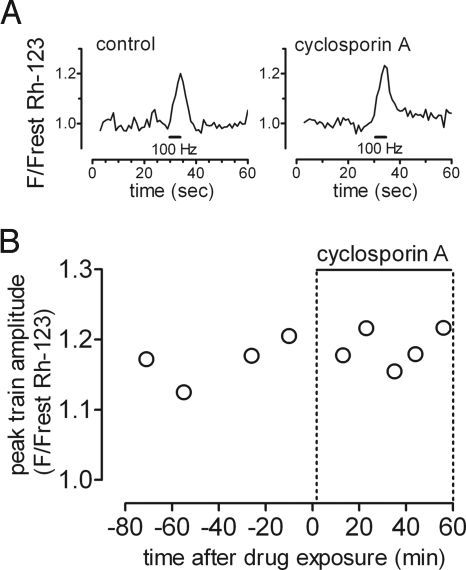
The electrical gradient across the mitochondrial inner membrane (Psi(m)) is established by electron transport chain (ETC) activity and permits mitochondrial Ca(2+) sequestration. Using rhodamine-123, we determined how repetitive nerve stimulation (100 Hz) affects Psi(m) in motor terminals innervating mouse levator auris muscles. Stimulation-induced Psi(m) depolarizations in wild-type (WT) terminals were small (<5 mV at 30 degrees C) and reversible. These depolarizations depended on Ca(2+) influx into motor terminals, as they were inhibited when P/Q-type Ca(2+) channels were blocked with omega-agatoxin. Stimulation-induced Psi(m) depolarization and elevation of cytosolic [Ca(2+)] both increased when complex I of the ETC was partially inhibited by low concentrations of rotenone (25-50 nmol/l). This finding is consistent with the hypothesis that acceleration of ETC proton extrusion normally limits the magnitude of Psi(m) depolarization during mitochondrial Ca(2+) uptake, thereby permitting continued Ca(2+) uptake. Compared with WT, stimulation-induced increases in rhodamine-123 fluorescence were approximately 5 times larger in motor terminals from presymptomatic mice expressing mutations of human superoxide dismutase I (SOD1) that cause familial amyotrophic lateral sclerosis (SOD1-G85R, which lacks dismutase activity; SOD1-G93A, which retains dismutase activity). Psi(m) depolarizations were not significantly altered by expression of WT human SOD1 or knockout of SOD1 or by inhibiting opening of the mitochondrial permeability transition pore with cyclosporin A. We suggest that an early functional consequence of the association of SOD1-G85R or SOD1-G93A with motoneuronal mitochondria is reduced capacity of the ETC to limit Ca(2+)-induced Psi(m) depolarization, and that this impairment contributes to disease progression in mutant SOD1 motor terminals.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Repetitive nerve stimulation transiently opens the mitochondrial permeability transition pore in motor nerve terminals of symptomatic mutant SOD1 mice.Neurobiol Dis. 2011 Jun;42(3):381-90. doi: 10.1016/j.nbd.2011.01.031. Epub 2011 Feb 18. Neurobiol Dis. 2011. PMID: 21310237 Free PMC article.
-
Stimulation-induced mitochondrial [Ca2+] elevations in mouse motor terminals: comparison of wild-type with SOD1-G93A.J Physiol. 2003 Jun 15;549(Pt 3):719-28. doi: 10.1113/jphysiol.2003.041905. Epub 2003 Apr 25. J Physiol. 2003. PMID: 12717010 Free PMC article.
-
Mitochondria in motor nerve terminals: function in health and in mutant superoxide dismutase 1 mouse models of familial ALS.J Bioenerg Biomembr. 2011 Dec;43(6):581-6. doi: 10.1007/s10863-011-9392-1. J Bioenerg Biomembr. 2011. PMID: 22089637 Free PMC article. Review.
-
Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention.Neurobiol Dis. 2011 Jun;42(3):265-75. doi: 10.1016/j.nbd.2011.01.016. Epub 2011 Feb 3. Neurobiol Dis. 2011. PMID: 21296666
-
Transgenic mice with human mutant genes causing Parkinson's disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration.Rev Neurosci. 2007;18(2):115-36. doi: 10.1515/revneuro.2007.18.2.115. Rev Neurosci. 2007. PMID: 17593875 Review.
Cited by
-
Dysregulated mitochondrial Ca2+ and ROS signaling in skeletal muscle of ALS mouse model.Arch Biochem Biophys. 2019 Mar 15;663:249-258. doi: 10.1016/j.abb.2019.01.024. Epub 2019 Jan 22. Arch Biochem Biophys. 2019. PMID: 30682329 Free PMC article. Review.
-
Insulin and Insulin-Sensitizing Drugs in Neurodegeneration: Mitochondria as Therapeutic Targets.Pharmaceuticals (Basel). 2009 Dec 23;2(3):250-286. doi: 10.3390/ph2030250. Pharmaceuticals (Basel). 2009. PMID: 27713238 Free PMC article. Review.
-
Mitochondrial pathobiology in ALS.J Bioenerg Biomembr. 2011 Dec;43(6):569-79. doi: 10.1007/s10863-011-9395-y. J Bioenerg Biomembr. 2011. PMID: 22083126 Free PMC article. Review.
-
Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca(2+) transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice.Front Cell Neurosci. 2014 Nov 14;8:353. doi: 10.3389/fncel.2014.00353. eCollection 2014. Front Cell Neurosci. 2014. PMID: 25452714 Free PMC article.
-
ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2.Hum Mol Genet. 2010 Aug 1;19(15):2974-86. doi: 10.1093/hmg/ddq202. Epub 2010 May 11. Hum Mol Genet. 2010. PMID: 20460269 Free PMC article.
References
-
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
