

Cooperative assembly and misfolding of CFTR domains in vivo
- PMID: 19176754
- PMCID: PMC2663924
- DOI: 10.1091/mbc.e08-09-0950
Cooperative assembly and misfolding of CFTR domains in vivo
Abstract
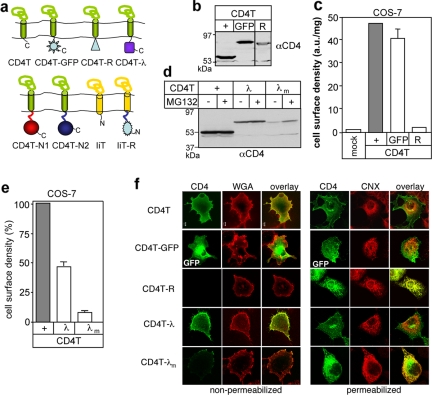
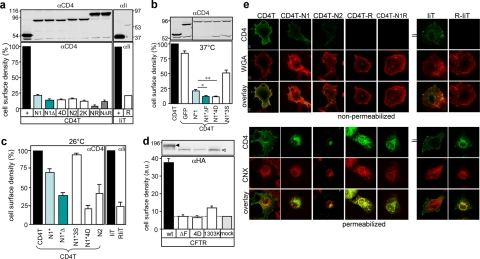
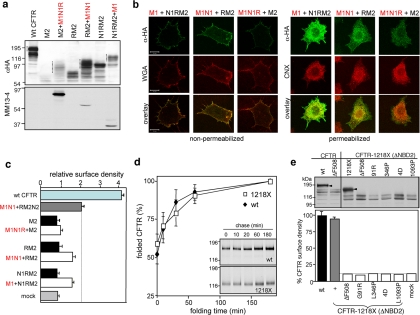
The cystic fibrosis transmembrane conductance regulator (CFTR) architecture consists of two membrane spanning domains (MSD1 and -2), two nucleotide binding domains (NBD1 and -2), and a regulatory (R) domain. Several point mutations lead to the channel misprocessing, with limited structural perturbation of the mutant domain. To gain more insight into the basis of CFTR folding defect, the contribution of domain-wise and cooperative domain folding was assessed by determining 1) the minimal domain combination that is recognized as native and can efficiently escape the endoplasmic reticulum (ER) retention and 2) the impact of mutation on the conformational coupling among domains. One-, two-, three-, and most of the four-domain assemblies were retained at the ER. Solubilization mutations, however, rescued the NBD1 processing defect conceivably by thermodynamic stabilization. The smallest folding unit that traversed the secretory pathway was composed of MSD1-NBD1-R-MSD2 as a linear or split polypeptide. Cystic fibrosis-causing missense mutations in the MSD1, NBD1, MSD2, and NBD2 caused conformational defect in multiple domains. We propose that cooperative posttranslational folding is required for domain stabilization and provides a plausible explanation for the global misfolding caused by point mutations dispersed along the full-length CFTR.
Figures




References
-
- Ashman J. B., Miller J. A role for the transmembrane domain in the trimerization of the MHC class II-associated invariant chain. J. Immunol. 1999;163:2704–2712. - PubMed
-
- Barriere H., Nemes C., Lechardeur D., Khan-Mohammad M., Fruh K., Lukacs G. L. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in mammalian cells. Traffic. 2006;7:282–297. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
